Das vollständige 16S-Gen bietet eine bessere taxonomische Auflösung
Das ~1500 bp 16S rRNA-Gen umfasst neun variable Regionen, die in der hochkonservierten 16S-Sequenz durchsetzt sind (Abb. 1a). Die Sequenzierung des gesamten Gens wurde ursprünglich durch Sanger-Sequenzierung durchgeführt., Dies erforderte das Klonen von Genen, das Erzeugen und Zusammenstellen von zwei bis drei Lesevorgängen pro Klon und das Erzeugen einer begrenzten Abtasttiefe bei hohen Kosten und Aufwand. Gegenwärtig sequenziert die überwiegende Mehrheit der Studien jedoch nur einen Teil des Gens, da die weit verbreitete Illumina-Sequenzierungsplattform (höherer Durchsatz, geringere Kosten, reduzierter Aufwand im Vergleich zu Sanger) kurze Sequenzen (≤300 Basen) erzeugt., Es werden daher verschiedene Teilregionen des Gens angestrebt, die von einzelnen variablen Regionen wie V4 oder V6 bis zu drei variablen Regionen wie V1–V3 oder V3–V5 reichen (die im Human Microbiome Project in Verbindung mit der Sequenzierungsplattform 454 verwendet werden9).
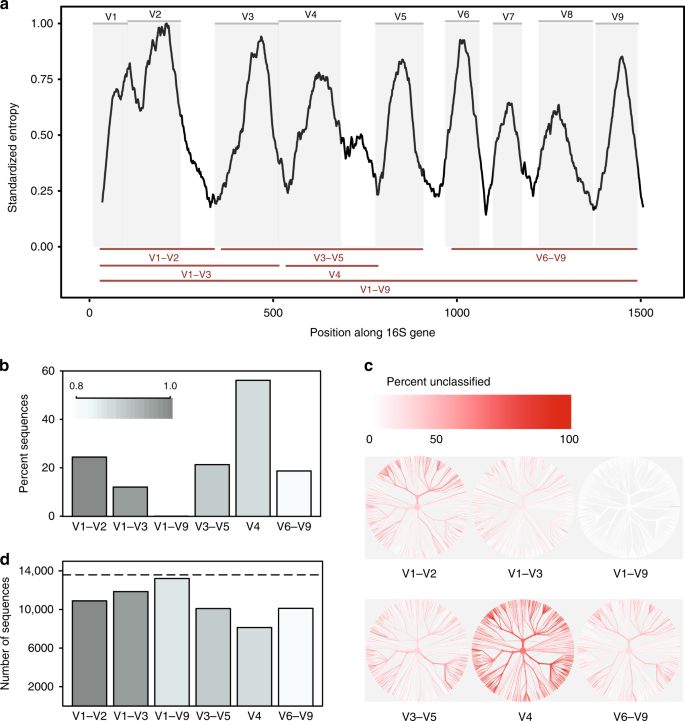
In-silico Vergleich der 16S-rRNA-variable Regionen. eine Shannon-Entropie über die 16S-gen auf der Grundlage der Ausrichtung eines einzigen repräsentativen Sequenz für jeden bekannten Arten, die in der Datenbank von Greengenes., Sequenzen wurden auf ein einzelnes Referenz-16S-Gen für Escherichia coli K-12 MG1655 ausgerichtet (NCBI-Gen ID 947777). Graue Paneele stellen variable Bereiche dar, die durch häufig verwendete Primerbindungsstellen definiert sind (ergänzende Tabelle 1). Variable Regionen, die in dieser Studie berücksichtigt werden, werden als rote Linien dargestellt (unten). b Anteil der Sequenzen für jede Variablenregion, die bei der Klassifizierung jeder Sequenz anhand der Referenzdatenbank, aus der sie abgeleitet wurde, bei einer Konfidenzschwelle von 80% nicht auf Artenebene identifiziert werden konnten (RDP-Klassifikator)., c-Bäume basierend auf der Taxonomie der in der In-Silico-Datenbank vorhandenen Sequenzen. Für jede variable Region wird derselbe Baum bereitgestellt. Die Farbe jedes Zweiges spiegelt den Anteil der Sequenzen innerhalb jeder Klade wider, die nicht auf Artenebene identifiziert werden konnten. d Die Anzahl der OTUs, die beim Clustern von Sequenzen für jeden Variablenbereich bei 99% Sequenzähnlichkeit erstellt wurden. Gestrichelte Linie gibt die Anzahl der eindeutigen Sequenzen (>1% anders) in der ursprünglichen Datenbank an., Quelldaten werden als Quelldatendatei bereitgestellt
Wir argumentieren, dass die Ausrichtung auf Unterregionen aufgrund technologischer Einschränkungen ein historischer Kompromiss darstellt10. Heute sind sowohl PacBio-als auch Oxford-Nanopore-Sequenzierungsplattformen in der Lage, routinemäßig Lesevorgänge von mehr als 1500 bp zu erzeugen, und die Hochdurchsatzsequenzierung des vollständigen 16S-Gens wird immer häufiger., Wir schlagen daher vor, dass die Rechtfertigung für diesen Kompromiss überarbeitet werden muss, und wir haben ein einfaches In-Silico-Experiment durchgeführt, um den Vorteil der Sequenzierung von 16S in voller Länge gegenüber dem Targeting von Teilregionen zu demonstrieren.
Wir haben eine Reihe von nicht redundanten (dh > 1% verschiedenen) 16S-Sequenzen in voller Länge aus einer öffentlichen Datenbank (Greengenes) heruntergeladen., Unter Ausnutzung der Tatsache, dass ein wesentlicher Teil dieser Sequenzen PCR-Primer-Bindungsstellen enthielt, trimmten wir sie, um In-Silico-Amplikons für verschiedene Teilregionen zu erzeugen, basierend auf der Position von PCR-Primern, die üblicherweise in Mikrobiomstudien verwendet werden (Abb. 1a und Ergänzende Tabellen 1-2)., Unter der Annahme, dass jede Sequenz in unserer heruntergeladenen Datenbank eine eindeutige Spezies darstellt, verwendeten wir dann einen Common Classification Approach (the Ribosome Database Project (RDP) classifier11), um die Häufigkeit zu berechnen, mit der In-Silico-Amplicons für jede Unterregion eine genaue taxonomische Klassifikation auf Artenebene liefern können (unter Verwendung der ursprünglichen Datenbank als Referenz). In einem zweiten Experiment, Wir gruppierten auch unsere In-Silico-Amplicons, um OTUs bei verschiedenen zu erzeugen, häufig verwendet, Sequenzähnlichkeitsschwellen (97%, 98%, 99%).,
Wir fanden heraus, dass sich Unterregionen wesentlich in dem Ausmaß unterschieden, in dem sie sicher zwischen den 16S-Sequenzen in voller Länge unterscheiden konnten, die zur Darstellung von Arten verwendet wurden (Abb. 1b). Die V4-Region schnitt am schlechtesten ab, wobei 56% der In-Silico-Amplicons auf dieser taxonomischen Ebene nicht sicher mit ihrer Herkunftsfolge übereinstimmten. Wenn dagegen eine Sequenz in voller Länge mit allen variablen Regionen verwendet wurde, war es möglich, fast alle Sequenzen als die richtige Spezies zu klassifizieren (Ergänzende Abb. 1a)., Die Änderung der Datenbanken und der Schwellenwerte für das Klassifikationskonfidenz beeinflussten den Anteil der In-Silico-Amplikons, die genau abgeglichen werden konnten, beeinflussten jedoch nicht die vorherrschenden Trends (Ergänzende Abb. 1a, b).
Zweitens zeigten verschiedene Teilregionen Verzerrungen in den bakteriellen Taxa, die sie identifizieren konnten (Abb. 1c). Zum Beispiel hat die V1–V2-Region bei der Klassifizierung von Sequenzen, die zu den Phylum-Proteobakterien gehören, eine schlechte Leistung erbracht, während die V3-V5-Region bei der Klassifizierung von Sequenzen, die zu den Phylum-Aktinobakterien gehören, eine schlechte Leistung erbracht hat (Ergänzende Abb. 2)., Ähnliche Trends wurden auf Genusebene für Taxa von potenzieller medizinischer Relevanz beobachtet. Obwohl die gesamte V1–V9-Region durchweg die besten Ergebnisse erzielte, war die V6–V9-Region insbesondere die beste Unterregion für die Klassifizierung von Sequenzen der Gattungen Clostridium und Staphylococcus, die V3–V5–Region führte zu guten Ergebnissen für Klebsiella und die V1-V3-Region zu guten Ergebnissen für Escherichia/Shigella (Ergänzende Abb. 2 und Quelldaten).
Schließlich wirkte sich die Wahl der Subregion dramatisch auf die Anzahl der OTUs aus, die beim Clustern von In-Silico-Amplikonen zur Erzeugung von OTUs gebildet wurden., Beim Clustering bei 99% Sequenzidentität konnten alle Unterregionen die Anzahl der verschiedenen Sequenzen in der ursprünglichen Datenbank nicht neu erstellen; Die V4-Region führte jedoch erneut die schlechteste Leistung aus (Abb. 1d). Insbesondere war die relative Anzahl der OTUs, die von jeder Unterregion erzeugt wurden, bei unterschiedlichen Identitätsschwellen nicht konsistent (97%, 98%, 99%, Ergänzende Abb. 3), was darauf hinweist, dass das Verhalten von Clustering-Algorithmen schwierig vorherzusagen sein kann, wenn die Menge an Informationen, die in einem sequenzierten Bereich enthalten sind, sehr variabel ist.,
Im Abschluss, targeting sub-Regionen repräsentiert einen historischen Kompromiss, das war ausreichend für die Identifizierung der taxa in die Gattung der Ebene oder oben. Unser einfaches In-Silico-Experiment zeigt jedoch, dass es nicht gültig ist anzunehmen, dass eine immer feinere Gruppierung dieser Teilregionen zu einer verbesserten taxonomischen Auflösung führt, die notwendig ist, um Arten widerzuspiegeln. Obwohl einige Teilregionen (z. B. V1–V3) eine angemessene Annäherung an die 16S-Vielfalt bieten, erfassen die meisten keine ausreichende Sequenzvariation, um zwischen eng verwandten Taxa zu unterscheiden., Wir stellen auch fest, dass diskriminierende Polymorphismen auf bestimmte variable Regionen beschränkt sein können; Daher sind bestimmte Unterregionen besser geeignet, um eng verwandte Mitglieder bestimmter Taxa zu diskriminieren.
16S Genkopiervarianten spiegeln Stamm-Level-Variation
Clustering von 16S-Sequenzen in OTUs hat historisch zwei Zwecke gedient. Erstens wurden geringfügige Artefaktsequenzvarianten aufgrund von PCR-Amplifikations-und Sequenzierungsfehlern beim Zusammenklappen von Sequenzen in Gruppen entfernt. Zweitens hat es legitime Sequenzvarianten zusammengebrochen, die zwischen eng verwandten bakteriellen Taxa existieren., Obwohl letzteres möglicherweise nicht immer wünschenswert ist, liegt es nahe, dass Sie nicht zwischen bakteriellen Taxa unterscheiden können, deren 16S-Sequenzen mit einer Rate variieren, die niedriger ist als der Fehler, der auf einer bestimmten Sequenzierungsplattform auftritt.
In jüngster Zeit haben Fortschritte in CCS die Fehlerraten von Sequenzierungsplattformen mit langer Lesezeit dramatisch verbessert. Gleichzeitig haben Rechenmethoden es ermöglicht, zwischen legitimen und artefaktmäßigen Sequenzvariationen zu unterscheiden., Diese technologischen und methodischen Fortschritte bedeuten, dass Forscher jetzt das Potenzial haben, Hochdurchsatzsequenzierungen durchzuführen, die Einzelnukleotidvarianten über das gesamte 16S-Gen genau nachweisen können.
Obwohl es verlockend ist anzunehmen, dass Einzelnukleotidvarianten unterschiedliche, eng verwandte Taxa darstellen können,warnen wir vor dieser allzu simplen Interpretation,da viele Bakteriengenome mehrere polymorphe Kopien des 16S-Gens enthalten 12, 13, 14., Wir führten die PacBio CCS-Sequenzierung einer 36-Spezies bakterielle Mock-Community durch (ergänzende Tabelle 3 und ergänzende Abb. 4) um (i) zu demonstrieren, dass die 16S-Sequenz vieler Bakterien zwischen Operonen innerhalb desselben Genoms variiert und (ii) dass die Hochdurchsatzsequenzierung ausreichend genau ist, um diese intragenomischen Unterschiede aufzulösen.
Wir haben PacBio 16S-Sequenzen in voller Länge auf eine Referenzdatenbank ausgerichtet, die eine einzelne repräsentative 16S-Sequenz für jedes Mitglied unserer Scheingemeinschaft enthält, und die Ausrichtungsstatistiken verwendet, um die Genauigkeit dieses Sequenzierungsansatzes zu bewerten., Vergleicht man die Anzahl der zur Erzeugung eines CCS verwendeten Durchläufe mit dem Auftreten von Einzelnukleotidsubstitutionen, so deuteten Insertionen und Löschungen darauf hin, dass zehn Durchgänge diese kombinierten Fehler auf eine Mindestfrequenz von 1.0% minimieren könnten (obwohl es bemerkenswert war, dass der minimal erreichbare Fehler zwischen den Sequenzierungsläufen variierte; Ergänzende Abb. 5). Wir haben jedoch ein Zusammentreffen von Löschfehlern mit den Standort-Homopolymerläufen in unseren Referenzsequenzen beobachtet (Ergänzende Abb., 6), die nicht nukleotidspezifisch war und durch die Länge des sequenzierten Homopolymers verschlimmert wurde (Ergänzende Abb. 7). Anschließend validierten wir Deletionen innerhalb des Escherichia coli 16S-Gens unter Verwendung der Illumina Whole Genome Shotgun (WGS) – Sequenzierung, die zeigte, dass nur eine der Deletionen, die in PacBio-Sequenzen auftraten, echt war (Ergänzende Abb. 8).,
In der Überzeugung, dass die CCS-Sequenzierung 16S-Lesevorgänge mit einer geringen Häufigkeit von Substitutionsfehlern erzeugen kann, haben wir als nächstes argumentiert, dass ein Teil der Substitutionsfehler innerhalb genau ausgerichteter Lesevorgänge Variationen widerspiegeln sollte, die auf 16S-Polymorphismen innerhalb des Genoms einer Spezies zurückzuführen sind12. Zum Beispiel liest ausgerichtet auf die E. coli Stamm K-12 substr. MG1655 zeigte ein Substitutionsprofil, das genau das widerspiegelte, was durch Ausrichten aller sieben der 16S-Sequenzen vorhergesagt wurde, von denen bekannt ist, dass sie in diesem Genom vorhanden sind15 (Abb. 2a, c)., Wir konnten die Stöchiometrie dieser Nukleotidsubstitutionen weiter validieren, indem wir Variationen in vergleichsweise ausgerichteten Illumina WGS-Lesevorgängen quantifizierten (Abb. 2b) und zeigen, dass ein ähnliches Substitutionsprofil über mehrere Sequenzierungsläufe hinweg reproduzierbar war (Ergänzende Abb. 9)., Ausrichtungen an anderen Referenzsequenzen in unserer Scheingemeinschaft zeigten einen ähnlichen Trend zu häufigen Substitutionen, die auf bestimmte Basispositionen entlang des 16S-Gens lokalisiert waren, obwohl wir feststellten, dass das Signal-Rausch-Verhältnis signifikant zunahm, wenn das betreffende 16S-Gen weniger als 100 ausgerichtete Lesevorgänge aufwies (Ergänzende Abb. 10).

Polymorphismen in E. coli 16S rRNA-gen-Sequenzen. a Die Position und Häufigkeit von Substitutionen, die in E auftreten., coli Stamm K-12 MG1655 V1-V9 Amplicons aus unserer Mock Community generiert und auf der PacBio RS II Plattform sequenziert. b Die Position und Häufigkeit von Substitutionen bei Lesevorgängen, die durch genomische Sequenzierung des isolierten E. coli-Stammes K-12 MG1655 auf der Illumina MiSeq-Plattform erzeugt werden. Vergrößerte Regionen zeigen jeweilige Positionen in der Ausrichtung aller sieben 16S-Gene, die im E. coli K-12 MG1655-Referenzgenom vorhanden sind. Die 16S-Sequenz aus dem rrnD-Operon ( * * ) wird als Referenz für alle SNP-Phasing verwendet. c Die vorhergesagte Nukleotid-substitution-Profil von E., coli K-12 MG1655 basierend auf der Ausrichtung der sieben im Referenzgenom vorhandenen 16S-Gensequenzen. d Das vorhergesagte Substitutionsprofil von E. coli O157 Sakai basierend auf der Ausrichtung der sieben im Referenzgenom vorhandenen 16S-Gensequenzen. Graue Paneele stellen variable Bereiche dar, die durch häufig verwendete Primerbindungsstellen definiert sind (ergänzende Tabelle 1). Gestrichelte Linien geben den erwarteten Anteil an Nukleotidsubstitutionen an, da sich in jedem Genom sieben 16S-Genkopien befinden., Quelldaten werden als Quelldatendatei bereitgestellt
Die Beobachtung, dass die Sequenzierung mit langer Lesezeit 16S-Polymorphismen innerhalb desselben Genoms identifizieren kann, hat wichtige Implikationen. Erstens zeigt es, dass es nicht gültig ist anzunehmen,dass Sequenzlesungen mit hohem Durchsatz, die sich um ein oder wenige Nukleotide unterscheiden, ein eindeutiges Taxa6, 16 darstellen. Innerhalb eines einzelnen Genoms können zwei oder mehr 16S-Sequenzen identisch sein, während andere eindeutig sein können., Entsprechend können einige homologe 16S-Loci eine identische Sequenz zwischen zwei eng verwandten Stämmen beibehalten, während andere an einer oder wenigen Nukleotidpositionen divergiert sein können. In diesem Zusammenhang sollte jede gemeinschaftsweite oder taxonomische Interpretation von 16S-Daten idealerweise die Tatsache berücksichtigen, dass die relative Häufigkeit von 16S-Sequenzen, die sich aus sehr eng verwandten Taxa ergeben, eine lineare Kombination von (i) der Häufigkeit widerspiegelt, mit der jede einzelne Sequenz über Genome hinweg dargestellt wird, und (ii) der relativen Häufigkeit der Genome für jedes Taxon.,
Zweitens, obwohl die intragenomische 16S-Sequenzvariation die Analyse auf Gemeinschaftsebene erschwert, hat sie auch das Potenzial, die Leistung des 16S-Gens zur Unterscheidung zwischen eng verwandten Taxa zu erhöhen, da sie einen sequenzbasierten Vergleich ermöglicht, der sich über mehrere divergente Loci erstreckt. Beispielsweise besteht eine ausreichende Nukleotidvariation, um E. coli-Stamm K-12 MG1655 vom enterohämorrhagischen Stamm O157 Sakai zu unterscheiden (Abb. 2c, d)., Daher argumentieren wir, dass, wenn angemessen berücksichtigt, mehrere polymorphe 16S Kopien sind keine Unannehmlichkeit zu übersehen, sondern sie ermöglichen die 16S-Gen in Stamm-Level-Mikrobiom-Analyse verwendet werden. Wir stellen auch fest, dass die Fähigkeit der intragenomischen 16S-Sequenzvariation, eng verwandte Taxa zu unterscheiden, wahrscheinlich abnimmt, wenn partielle 16S-Sequenzen verwendet werden. Zum Beispiel unterscheiden SNPs die E. coli-Stämme K-12 MG1655 (Abb. 2c) aus O157 Sakai (Abb. 2d) sind in variablen Regionen V1, V2, V6 und V9 zu finden.,
16S Polymorphismen können in vivo aufgelöst werden
Mikrobiomgemeinschaften sind oft komplex und existieren in verschiedenen biochemischen Umgebungen (z. B. Stuhl, Speichel, Sputum usw.) und enthält viele Hundert einzigartige Taxa, deren relative Fülle einen breiten Dynamikbereich umfasst. Diese Komplexität ist weder in In-Silico-noch in Mock-Community-Experimenten gut vertreten. Wir führten daher ein zusätzliches Experiment durch, um zu zeigen, dass die Sequenzierung des vollständigen 16S-Gens unter Berücksichtigung intragenomischer 16S-SNPs eng verwandte bakterielle Taxa in vivo auflösen kann.,
Wir führten die PacBio CCS-Sequenzierung der V1-V9-Region für vier menschliche Stuhlproben durch, die von gesunden erwachsenen Freiwilligen gesammelt wurden. Zum Vergleich haben wir die V1–V3-Region mit dem Illumina MiSeq sequenziert und, um einen Benchmark für die taxonomische Quantifizierung auf Artenebene zu liefern, metagenomische WGS (mWGS)-Sequenzierung mit dem Illumina NextSeq durchgeführt. Um zu bewerten, inwieweit jeder dieser Sequenzierungsansätze eng verwandte Taxa auflösen kann, konzentrierten wir uns auf die Gattung Bacteroides., Diese Gattung ist nicht nur im menschlichen Darm reichlich vorhanden, sondern auch sehr vielfältig und enthält mehrere Arten, die sowohl gute als auch schlechte Auswirkungen auf die menschliche Gesundheit ausüben können17. Es wurde auch früher als Modelltaxon verwendet, um die Nützlichkeit des 16S-Gens für hochauflösende taxonomische Analyse18 zu demonstrieren.
Wenn wir Bacteroides Fülle auf der Genusebene berechnet, V1-V9 Sequenzierung und V1-V3 Sequenzierung erzeugt vergleichbare Ergebnisse., Beide Ansätze identifizierten zwei Personen mit geringer relativer Häufigkeit von Bacteroiden (~10-25%) und zwei Personen mit hoher relativer Häufigkeit von Bacteroiden (~40-60%; Abb. 3a). Die Quantifizierung auf Artenebene über mWGS-Sequenzierung ergab jedoch eine weitaus größere Vielfalt, wobei eine andere Bacteroides-Spezies im Darm jedes Individuums dominant war (Abb. 3b und Ergänzende Daten 1). Beim Clustern von OTUs mit 99% Identität konnten sowohl die V1–V9–als auch die V1-V3-Sequenzierung diese Variation auf Speziesebene widerspiegeln (Abb., 3b), mit der bemerkenswerten Ausnahme, dass die V1–V3-Sequenzierung Bacteroides intestinalis nicht nachwies, was in einer der vier menschlichen Darmmikrobiomproben reichlich vorhanden war. Basierend auf diesen Ergebnissen schließen wir, dass OTU-basierte Ansätze in Verbindung mit einer geeigneten Identitätsschwelle (z. B. 99%) das Potenzial haben, die im menschlichen Darm beobachtete Artenvielfalt aufzulösen. Wir stellen ferner fest, dass, obwohl die Sequenzierung von 16S in voller Länge für die Analyse auf Artenebene optimal sein kann, auch sehr informative variable Regionen (z. B. V1-V3) für diesen Zweck ausreichend sein können.,

die Erkennung von Bacteroides in menschlichen Stuhlproben. a Die relative Häufigkeit der Gattung Bacteroides in vier menschlichen Stuhlproben quantifiziert entweder V1–V9 Amplicons (x-Achse) oder V1–V3 Amplicons (y-Achse). b Die relative Häufigkeit von Bacteroides Spezies in den gleichen vier Proben. Die Artenfülle wurde aus mWGS–Sequenzierung oder aus V1–V3/V1-V9 OTUs quantifiziert, die bei 99% Identität erzeugt wurden., Die Häufigkeit wird für die am häufigsten vorkommenden Arten nach mWGS quantifiziert (für Abundanzschätzungen aller von jeder Plattform nachgewiesenen Bacteroides-Arten siehe ergänzende Tabelle 5). c-Nukleotidsubstitutionsprofile, die durch Ausrichten aller V1–V9-Amplikonsequenzen erzeugt werden, die der einzelnen OTU zugeordnet sind, die als Bacteroides vulgatus identifiziert wurde. Profile angezeigt, für die zwei Stuhlproben mit hoher B. vulgatus relative Häufigkeit (IronHorse und Scott). d Nukleotidsubstitutionsprofile, die aus den Referenzgenomen zweier verschiedener B. vulgatus-Stämme ATCC 848239 und mpk40 vorhergesagt wurden., Sowohl in c als auch in d wurden Nukleotidsubstitutionen relativ zu einem einzelnen Referenz-16S-Gen für B. vulgatus ATCC 8482 identifiziert (NCBI-Gen ID 5304800). Graue Paneele stellen variable Bereiche dar, die durch häufig verwendete Primerbindungsstellen definiert sind (ergänzende Tabelle 1). Gestrichelte Linien geben den erwarteten Anteil an Nukleotidsubstitutionen an, da sich in jedem Genom sieben 16S-Genkopien befinden., Quelldaten werden als Quelldatendatei bereitgestellt
Unter Ausnutzung der Tatsache, dass Bacteroides vulgatus in zwei unserer menschlichen Darmmikrobiomproben in hoher relativer Häufigkeit vorhanden war, fragten wir als nächstes, ob intragenomische Variationen zwischen 16S-Genkopien in vivo nachgewiesen werden könnten. Wir haben jede Sequenz in voller Länge ausgerichtet, die als zu unserem B. vulgatus V1–V9 OTUs gehört (Abb. 3b und ergänzende Daten 1) zu einer einzigen repräsentativen B. vulgatus 16S Gensequenz. Wir haben dann die resultierenden Nukleotidsubstitutionsprofile verglichen (Abb., 3c) mit Profilen, die aus zwei in der NCBI RefSeq-Datenbank vorhandenen Referenzgenomen vorhergesagt werden19 (Abb. dreidimensional).
Die Mehrzahl der Nukleotidvariation, die in unserem in vivo erzeugten B. vulgatus OTU vorhanden ist, spiegelt die wahre Variation wider, die auf intragenomische Polymorphismen zurückzuführen ist. Im Gegensatz dazu erschien die Variation wahrscheinlich aufgrund von Sequenzierungsfehlern niedrig und deutlich unter der minimalen ~14% – Frequenz, die erwartet würde, wenn in jeder Probe ein einzelner B. vulgatus-Stamm mit sieben 16S-Genkopien in seinem Genom vorhanden wäre (Abb. 3c, gestrichelte Linien).
Obwohl wir die wahre Anzahl von B nicht kannten., vulgatusstämme, die in jeder In-vivo-Probe vorhanden waren, Es war bemerkenswert, dass beide Nukleotidsubstitutionsprofile eine engere Ähnlichkeit mit dem Stamm ATCC 8482 hatten als mpk. Variation gab es auch an bestimmten Loci, die möglicherweise auf aussagekräftige Unterschiede zwischen dem In vivo-und ATCC 8482-Referenzgenom hinweisen könnten. Zum Beispiel wurde ein einzelner Polymorphismus im V5-Bereich von ATCC 8482 nachgewiesen, der in drei 16S-Kopien (43%) vorhanden war. In der ersten In-vivo-Probe (Scott) war dieser Polymorphismus in 84% der Lesevorgänge vorhanden, während er in der zweiten (IronHorse) in 69% der Lesevorgänge vorhanden war., Diese Zahlen entsprechen eng den erwarteten Zahlen, wenn ein Polymorphismus sechs bzw. fünf von sieben 16S-Genen vorhanden wären.
Zusammenfassend zeigen wir, dass die 16S-Sequenzierung des menschlichen Darmmikrobioms in voller Länge Einzelnukleotidsubstitutionen genau auflösen kann, die die intragenomische Variation zwischen 16S-Genkopien widerspiegeln. Das Vorhandensein einer solchen Variation zeigt an, dass 16S-Sequenzen gruppiert werden müssen, um aussagekräftige taxonomische Einheiten widerzuspiegeln., Mit OTUs, die bei 99% Identität gruppiert sind, zeigen wir, dass 16S in voller Länge das Potenzial haben, Arten und sogar taxonomische Auflösung auf Stammebene bereitzustellen. Die Analyse der mikrobiellen Gemeinschaften auf diesen taxonomischen Ebenen verspricht eine ganz andere Perspektive als die von Genus-Level-Abundanzschätzungen.
Intragenomische 16S-Polymorphismen sind weit verbreitet
Nachdem gezeigt wurde, dass es möglich ist, intragenomische Kopiervarianten in vivo aufzulösen, versuchten wir als nächstes festzustellen, inwieweit solche Kopiervarianten in Taxa vorkommen, die üblicherweise im menschlichen Darmmikrobiom vorkommen., Wir haben weiter versucht festzustellen, ob solche Profile routinemäßig zur Unterscheidung zwischen Stämmen derselben Spezies verwendet werden können.
Wir kultivierten 381 Taxa aus dem Darmmikrobiom der in Abb. 3, sowie von anderen Personen, die an derselben ursprünglichen Studie20 teilnehmen (ergänzende Daten 2). Anschließend führten wir eine 16S-Gensequenzierung in voller Länge an Isolaten und ausgerichteten sequenzierten Lesevorgängen durch, um Nukleotidsubstitutionen zu identifizieren, die für intragenomische 16S-Genkopievarianten charakteristisch sind.,
Die taxonomische Klassifikation der Isolate identifizierte 58 mutmaßliche Arten (ergänzende Daten 2), während die Gruppierung einer einzelnen repräsentativen Sequenz für jedes Isolat bei 99% Ähnlichkeit zu 61 OTUs führte (wobei jeder OTU zwischen 1 und 73 Isolate zugewiesen waren). Insgesamt hatten 349 von 381 sequenzierten Isolaten (54 von 61 OTUs) einen oder mehrere SNP, was auf das Vorhandensein von 16S-Genpolymorphismen hinweist, und 205 eindeutige SNP-Profile wurden identifiziert, wenn ein potenzieller Sequenzierungsfehler berücksichtigt wurde (Abb. 4a und ergänzende Angaben 2).

Intragenomische 16S-Genpolymorphismen in menschlichen Darmmikrobiomisolaten. ein Ort von SNPs, der in den 16S-Genen individuell kultivierter bakterieller Isolate vorhanden ist. SNP-Standorte wurden durch phasenweise 16S-Gensequenzen in voller Länge identifiziert, die für jedes einzelne Isolat generiert wurden. Die X-Achse bezeichnet die Position entlang des 16S-Gens. Die Y-Achse bezeichnet einzelne Isolate, die basierend auf ihrer abgeleiteten Phylogenie gruppiert sind. Dunkelblauer Bereich zeigt den Ort eines Polymorphismus an., Zur Verdeutlichung werden maximal fünf Isolate derselben Art gezeigt. Einzelheiten zu Nukleotidsubstitutionsprofilen für alle sequenzierten Isolate finden Sie in den ergänzenden Daten 2. b-d Beispiele für Nukleotidsubstitutionsprofile, die Unterschiede auf Stammebene zwischen Isolaten zeigen, die zu drei Bakterienarten gehören: b Shigella flexneri; c Bifidobacterium longum; d Collinsella aerofaciens. Für jede Spezies werden zwei Isolat-Nukleotid-Substitutionsprofile gezeigt; zusätzliche Beispiele finden sich jedoch in ergänzenden Daten 2., Isolate wurden als zu derselben Spezies gehörend identifiziert, wenn ihre repräsentativen Sequenzen derselben OTU zugewiesen wurden, wenn sie bei 99% Sequenzidentität gruppiert wurden. Die taxonomische Identifizierung wurde unter Verwendung von BLAST durchgeführt, um repräsentative Sequenzen an der NCBI 16S BLAST-Datenbank auszurichten (siehe Methoden). Graue Paneele stellen variable Bereiche dar, die durch häufig verwendete Primerbindungsstellen definiert sind (ergänzende Tabelle 1). Gestrichelte Linien geben den erwarteten Anteil an Nukleotidsubstitutionen an, wenn die Anzahl der 16S-Genkopien für jedes Genom vorhergesagt wird., Quelldaten werden als Quelldatendatei bereitgestellt
Insbesondere der Vergleich von SNP-Profilen für Isolate, die derselben OTU zugewiesen wurden, ergab häufig Unterschiede in der Häufigkeit von SNPs, die auf Unterschiede in intragenomischen 16S-Genkopien zwischen eng verwandten Taxa hindeuteten. Beispiele für verschiedene Substitutionsprofile sind für drei Taxa dargestellt (Abb. 4b-d), die auf eine Dehnungsschwankung hindeuten, vergleichbar mit der, die wir prinzipiell für E. coli demonstriert haben (Abb. 2b).,
Zusammenfassend zeigen wir, dass viele der kultivierbaren Mitglieder des menschlichen Darmmikrobioms häufig 16S-Genpolymorphismen besitzen, die, wenn sie richtig berücksichtigt werden, das Potenzial haben, Stämme derselben Spezies aufzulösen.