plné 16S gen poskytuje lepší taxonomické rozlišení
~1500 bp 16S rRNA genu se skládá z devíti variabilní regiony proložen vysoce konzervované sekvence 16S (Obr. 1a). Sekvenování celého genu bylo původně provedeno sekvenováním Sanger., To vyžadovalo klonování genů, generování a sestavení dvou až tří čtení na klon, a produkovat omezenou hloubku odběru vzorků za vysoké náklady a úsilí. V současné době, nicméně, drtivá většina studií sekvence pouze část genu, protože široce používán Illumina sekvenování platformy (vyšší propustnost, nižší náklady, snížení úsilí ve srovnání s Sanger) produkuje krátké sekvence ( ≤ 300 bází)., Různých sub-regionech genu jsou tedy cílené, od jedné proměnné regionů, jako V4 nebo V6, pro tři proměnné regiony, jako je například V1–V3 nebo V3–V5 (používá se v Human Microbiome Project, ve spojení s 454 sekvenování platform9).
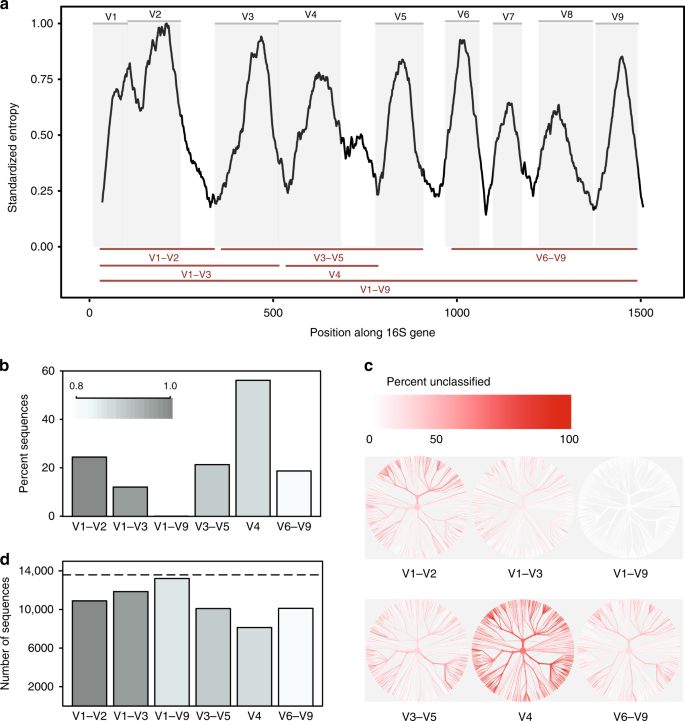
in-silico srovnání proměnných oblastí 16S rRNA. Shannonova entropie napříč genem 16S založená na zarovnání jediné reprezentativní sekvence pro každý známý druh přítomný v databázi Greengenes., Sekvence byly zarovnány proti jedinému referenčnímu genu 16s pro Escherichia coli K-12 MG1655 (NCBI GEN ID 94777). Šedé panely zobrazují variabilní oblasti definované běžně používanými místy pro vázání primerů (doplňková Tabulka 1). Variabilní oblasti zvažované v této studii jsou zobrazeny jako červené čáry (dole). B podíl sekvencí pro každou proměnnou oblast, která nemohla být identifikována na úrovni druhů při klasifikaci každé sekvence proti referenční databázi, ze které byla odvozena, s prahem spolehlivosti 80% (klasifikátor RDP)., c stromy založené na taxonomii sekvencí přítomných v databázi in-silico. Stejný strom je k dispozici pro každou proměnnou oblast. Barva každé větve odráží podíl sekvencí v každé clade, které nemohly být identifikovány na úrovni druhů. d počet Otu vytvořených při shlukování sekvencí pro každou proměnnou oblast při 99% sekvenční podobnosti. Přerušovaná čára označuje počet jedinečných sekvencí (> 1% různých) v původní databázi., Zdrojem dat jsou poskytovány jako Zdrojový Datový soubor.
tvrdí, že cílení sub-regionů představuje historický kompromis, vzhledem k technologii restrictions10. Dnes jsou jak pacbio, tak Oxfordské sekvenční platformy Nanopore schopny rutinně produkovat čtení přesahující 1500 bp a sekvenování plného 16S genu s vysokou propustností je stále častější., Proto jsme naznačují, že důvod pro tento kompromis musí být znovu a provedli jsme jednoduchý in-silico experiment k prokázání výhod full-délka 16S sekvenování přes zaměření sub-regionů.
stáhli jsme sadu non-redundantní (tj., > 1% odlišný), full-length 16S sekvence z veřejné databáze (Greengenes)., Využít skutečnosti, že podstatná část těchto sekvencí začleněna PCR primer-vazebná místa, jsme zdobené, aby generovat in-silico amplikony pro různé sub-regionů, na základě umístění PCR primery běžně používané v mikrobiomu studií (Obr. 1a a doplňkové tabulky 1-2)., Za předpokladu, že každá posloupnost v naší stažené databáze představuje unikátní druh, jsme pak použili společné klasifikace přístupu (Ribozomu Databáze Project (RDP) classifier11) pro výpočet frekvence, s níž in-silico amplikony pro každý sub-region by mohl poskytovat přesné, druhy-úroveň taxonomické klasifikace (použitím původní databáze jako referenční). V druhém experimentu jsme také seskupený naše in-silico amplikony generovat OTUs v různých, běžně používaných, sekvenční podobnost prahy (97%, 98%, 99%).,
zjistili jsme, že dílčí oblasti se podstatně lišily v rozsahu, v jakém by mohly s jistotou rozlišovat mezi sekvencemi 16S v plné délce používanými k reprezentaci druhů (obr. 1b). V4 regionu provádí nejhorší, s 56% in silico amplikony nedaří s jistotou odpovídají jejich sekvenci původu na této taxonomické úrovni. Naproti tomu, když v plné délce sekvence s proměnnou regionů byl použit, bylo možné klasifikovat téměř všechny sekvence jako správný druhů (Doplňkový Obr. 1a)., Změna databází a prahových hodnot spolehlivosti klasifikace ovlivnila podíl amplikonů in-silico, které by mohly být přesně porovnány, ale neovlivnily převládající trendy (doplňující obr. 1a, b).
za druhé, různé dílčí oblasti vykazovaly zaujatost v bakteriálních taxonech, které byly schopny identifikovat (obr. 1c). Například, V1–V2 regionu provádí špatně v klasifikaci sekvencí patřící do kmene Proteobacteria, vzhledem k tomu, že V3–V5 regionu provádí špatně v klasifikaci sekvencí patřící do kmene Actinobacteria (Doplňkový Obr. 2)., Podobné trendy byly pozorovány na úrovni rodu pro taxony potenciálního lékařského významu. I když full V1–V9 kraj trvale nejlepší výsledky, V6–V9 kraj byl zejména nejlepší sub-region pro klasifikaci sekvencí rodů Clostridium a Staphylococcus, V3–V5 regionu přinesla dobré výsledky pro Klebsiella, a V1–V3 regionu přinesla dobré výsledky pro Escherichia/Shigella (Doplňkový Obr. 2 a zdrojová Data).
a Konečně, výběr sub-regionu výrazně ovlivnily počet OTUs tvořen, když clustering in silico amplikony vytvořit OTUs., Když clustering na 99% sekvenční identity, všechny sub-regiony se nepodařilo obnovit počet odlišných sekvencí přítomných v původní databáze; nicméně, v regionu V4 se opět provádí nejhorší (Obr. 1d). Zejména, relativní počet Otu produkovaných každou podoblastí nebyl konzistentní při různých prahových hodnotách identity(97%, 98%, 99%, Doplňkový obr. 3), což naznačuje, že chování clustering algoritmů může být obtížné předvídat, kdy množství informace obsažené v sekvenované oblasti je velmi variabilní.,
Závěrem lze říci, že cílení subregionů představuje historický kompromis, který byl dostatečný pro identifikaci taxonů na úrovni rodu nebo vyšší. Náš jednoduchý experiment in-silico však ukazuje, že není platné předpokládat, že stále jemnější shlukování těchto subregionů povede ke zlepšení taxonomického rozlišení nezbytného k odrážení druhů. Ačkoli některé subregiony (např. V1-V3) poskytují přiměřenou aproximaci rozmanitosti 16S, většina nezachycuje dostatečnou sekvenční variaci, aby rozlišovala mezi úzce souvisejícími taxony., Upozorňujeme také, že diskriminační polymorfismy mohou být omezeny na konkrétní variabilní oblasti; některé subregiony tak budou vhodnější pro diskriminaci úzce příbuzných členů určitých taxonů.
varianty kopírování genů 16S odrážejí variaci na úrovni kmene
shlukování sekvencí 16S do Otu historicky sloužilo dvěma účelům. Nejprve odstranil drobné varianty artifactual sekvence kvůli chybám PCR amplifikace a sekvenování při kolapsu sekvencí do skupin. Za druhé, zhroutila legitimní varianty sekvencí, které existují mezi úzce souvisejícími bakteriálními taxony., I když toto nemusí být vždy žádoucí, je logické, že nelze rozlišovat mezi bakteriální taxony, jejichž sekvence 16S liší v míře, která je nižší než chyba s nimiž se setkávají na konkrétní sekvenační platformu.
v poslední době pokroky v CCS dramaticky zlepšily chybovost dlouho čtených sekvenčních platforem. Zároveň výpočetní metody umožnily rozlišovat mezi legitimní vs. artifactual variací sekvence., Tyto technologické a metodické pokroky znamenají, vědci mají nyní potenciál k provádění vysoce paralelních měrení, celogenomové sekvenování, že může přesně detekovat single-nukleotidové varianty v celé 16S gen.
Ačkoli je lákavé předpokládat, že single-nukleotidové varianty mohou představovat odlišné, úzce související taxony, varujeme proti tomu příliš zjednodušující interpretace vzhledem k tomu, že mnoho bakteriální genomy obsahují více polymorfních kopií 16S gene12,13,14., Provedli jsme pacbio CCS sekvenování 36 druhů bakteriální mock komunity (doplňková Tabulka 3 a doplňkový Obr. 4) doložit, že (i) sekvence 16S mnoha bakterií se pohybuje mezi operons v rámci téhož genomu a (ii) že high-throughput sekvenování je dostatečně přesné, aby řešení těchto intragenomic rozdíly.
Jsme vyrovnané PacBio full-délka 16 sekvencí na referenční databáze obsahuje jeden reprezentativní sekvence pro 16S každý člen našeho mock společenství a používá zarovnání statistiky vyhodnotit správnost těchto sekvenční přístup., Porovnání počtu průchodů slouží k vytvoření CCS s výskytem single-nukleotidové substituce, inzerce a delece uvedla, že deset prochází mohli minimalizovat tyto chyby v kombinaci na minimální frekvence < 1.0% (i když to bylo pozoruhodné, že minimální dosažitelná chyba se pohybovala mezi sekvenování běží; Doplňující Obr. 5). Nicméně, pozorovali jsme, náhoda odstranění chyb s umístěním homopolymer běží v našich referenčních sekvencí (Doplňkový Obr., 6), která nebyla specifická pro nukleotidy a byla zhoršena délkou sekvenovaného homopolymeru (Doplňkový obr. 7). My následně ověřena delece v Escherichia coli 16S gen pomocí Illumina whole genome shotgun (WGS) sekvenování, které prokázaly, že pouze jeden z delece vyskytující se v PacBio sekvence byla originální (Doplňkový Obr. 8).,
přesvědčen, že CCS sekvenování může produkovat 16 čte s nízkou frekvencí střídání chyby, dále jsme si řekli, že podíl substituce chyby v přesně v souladu čte, by měla odrážet rozdíly připadající na 16 polymorfismů v rámci druhu genome12. Například čte zarovnané s kmenem E. coli K-12 substr. MG1655 ukázal substituční profil, který odrážel přesně to, co předpovídalo zarovnání všech sedmi sekvencí 16S, o nichž je známo, že jsou přítomny v tomto genome15 (obr. 2a, c)., Dále jsme byli schopni ověřit stechiometrii těchto substitucí nukleotidů kvantifikací variací srovnatelně zarovnaných Illumina WGS (obr. 2b) a prokázat, že podobný substituční profil byl reprodukovatelný přes více sekvenačních běhů(Doplňkový obr. 9)., Zarovnání na jiné referenční sekvence, v našem mock společenství vykazovaly podobný trend bohatá substituce lokalizované na konkrétní základní pozice podél 16S gen, i když jsme na vědomí, že signál-šum se výrazně zvýšil, když 16S gen v otázce měl méně než 100 souladu čte (Doplňkový Obr. 10).

polymorfismy v genových sekvencích E.coli 16S rRNA. a postavení a četnost substitucí uvedených v e., coli kmen K–12 mg1655 v1-v9 amplikony generované z naší falešné komunity a sekvenované na platformě PacBio RS II. b polohy a frekvence substitucí v přečte vytvořené z genomové sekvenování izolovaných kmene E. coli K-12 MG1655 na Illumina MiSeq platformy. Zvětšené oblasti vykazují příslušné polohy v zarovnání všech sedmi genů 16S přítomných v referenčním genomu E.coli K-12 MG1655. 16S sekvence z rrnd operon ( * * ) se používá jako reference pro všechny SNP fázování. c předpokládaný substituční profil nukleotidu e., coli K – 12 MG1655 na základě zarovnání sedmi genových sekvencí 16S přítomných v referenčním genomu. d předpokládaný substituční profil E. coli O157 Sakai založený na zarovnání sedmi genových sekvencí 16S přítomných v referenčním genomu. Šedé panely zobrazují variabilní oblasti definované běžně používanými místy pro vázání primerů (doplňková Tabulka 1). Přerušované čáry ukazují očekávaný podíl substitucí nukleotidů, vzhledem k tomu, že v každém genomu je sedm kopií genů 16S., Zdrojem dat jsou poskytovány jako Zdrojový Datový soubor.
pozorování, že dlouhodobé číst sekvenování může identifikovat 16 polymorfismů v rámci téhož genomu má významné důsledky. Za prvé,ukazuje, že není platné předpokládat, že sekvence s vysokou propustností se liší jedním nebo několika nukleotidy představují zřetelný taxa6, 16. V rámci jednoho genomu mohou být dvě nebo více sekvencí 16S identické, zatímco jiné mohou být jedinečné., Odpovídajícím způsobem, některé homologní 16S loci mohou zachovat identickou sekvenci mezi dvěma úzce souvisejícími kmeny, zatímco jiné se mohou lišit v jedné nebo několika pozicích nukleotidů. V této souvislosti, jakékoli úrovni společenství nebo taxonomické interpretace 16S údaje by měly v ideálním případě v úvahu skutečnost, že relativní četnosti sekvencí 16S vyplývající z velmi blízce příbuzné taxony, které bude odrážet lineární kombinace (i) frekvence, s níž každý unikátní sekvence je zastoupena v celé genomy a (ii) relativní četnost genomů pro každý taxon.,
za Druhé, ačkoli intragenomic sekvence 16S změna komplikuje společenství-analýza na úrovni, to také má potenciál ke zvýšení výkonu 16S gen rozlišovat mezi blízce příbuzných taxonů, protože to umožňuje sekvence založené na srovnání prodloužit na více odlišných lokusů. Například existuje dostatečná variace nukleotidů, aby se odlišil kmen E. coli K-12 MG1655 od enterohemoragického kmene O157 Sakai (obr. 2c, d)., Tvrdíme tedy, že pokud jsou vhodně účtovány, vícenásobné polymorfní kopie 16S nejsou nepříjemností, které je třeba přehlédnout, spíše umožní použití genu 16S v analýze mikrobiomů na úrovni kmene. Také si všimneme, že síla intragenomické variace sekvence 16S k diskriminaci úzce souvisejících taxonů se pravděpodobně sníží, když se použijí částečné sekvence 16S. Například SNP rozlišující kmeny E. coli K-12 MG1655 (obr. 2c) od O157 Sakai (obr. 2D) se nacházejí v proměnných oblastech V1, V2, V6 a v9.,
16S polymorfismy lze vyřešit in vivo
mikrobiomové komunity jsou často složité, existující v různých biochemických prostředích (např.) a obsahuje mnoho stovek unikátních taxonů, jejichž relativní hojnost pokrývá široký dynamický rozsah. Tato složitost není dobře zastoupena ani v silico ani v mock komunitních experimentech. Proto jsme provedli další experiment, abychom prokázali, že sekvenování plného genu 16S při účtování intragenomických SNP 16s může vyřešit úzce související bakteriální taxony in vivo.,
Provedli jsme sekvenování PacBio CCS v oblasti v1 – v9 pro čtyři vzorky lidské stolice odebrané od zdravých dospělých dobrovolníků. Pro srovnání jsme zařadili V1–V3 regionu pomocí Illumina MiSeq a, poskytnout měřítko pro species-taxonomické úrovni kvantifikace, jsme provedli metagenomic WGS (mWGS) sekvenování pomocí Illumina NextSeq. Abychom zhodnotili, do jaké míry může každý z těchto sekvenačních přístupů vyřešit úzce související taxony, zaměřili jsme se na rod Bacteroides., Kromě toho, že je hojný v lidském střevě, je tento rod velmi rozmanitý a obsahuje více druhů, které mohou mít dobré i špatné účinky na lidské zdraví17. Dříve byl také používán jako vzorový taxon pro prokázání užitečnosti genu 16S pro taxonomickou analýzu s vysokým rozlišením 18.
Když jsme vypočítali množství Bacteroidů na úrovni rodu, sekvenování V1–v9 a sekvenování V1–V3 přinesly srovnatelné výsledky., Oba přístupy identifikovaly dva jedince s nízkou Bacteroides relativní hojnosti (~10-25%) a dva jedinci s vysokou Bacteroides relativní hojnosti (~40-60%; Obr. 3a). Kvantifikace na úrovni druhů pomocí sekvenování mwgs však odhalila mnohem větší rozmanitost ,přičemž ve střevě každého jednotlivce dominují různé druhy Bacteroidů (obr. 3b a doplňující údaje 1). Při shlukování Otu na 99% identity byly sekvenování v1–v9 i V1–V3 schopny odrážet tuto variaci na úrovni druhů (obr., 3b), s výjimkou, že V1–V3 sekvenování nezjistil Bacteroides intestinalis, který byl hojný v jednom ze čtyř lidského střevního mikrobiomu vzorků. Na základě těchto výsledků jsme dospěli k závěru, že při použití ve spojení s vhodným prahem identity (např. 99%) mají přístupy založené na OTU potenciál vyřešit rozmanitost na úrovni druhů pozorovanou v lidském střevě. Dále poznamenáváme, že i když sekvenování v plné délce 16S může být optimální pro analýzu na úrovni druhů, mohou být pro tento účel vhodné i vysoce informativní variabilní oblasti (např. V1-V3).,

Detekce Bacteroides v lidské stolici. a relativní hojnost rodu Bacteroides ve čtyřech vzorcích lidské stolice kvantifikovaných pomocí amplikonů v1-V9 (osa x) nebo amplikonů V1-V3 (osa y). b relativní hojnost druhů Bacteroides ve stejných čtyřech vzorcích. Hojnost druhů byla kvantifikována z sekvenování mWGS nebo z Otu V1–V3/v1-V9 generovaného na 99% identity., Hojnost je zobrazena u nejhojnějších druhů kvantifikovaných mWGS (pro odhady hojnosti všech druhů Bacteroidů zjištěných každou platformou, viz doplňková Tabulka 5). c Nukleotidové substituce profily vytvořené spojením všech V1–V9 sekvence amplikonu přiřazen jeden OTU identifikován jako Bacteroides vulgatus. Profily jsou zobrazeny pro dva vzorky stolice s vysokou relativní hojností B. vulgatus (IronHorse a Scott). d nukleotidové substituční profily předpovídané z referenčních genomů dvou různých kmenů B. vulgatus ATCC 848239 a mpk40., V obou c A d byly nukleotidové substituce identifikovány vzhledem k jedinému referenčnímu genu 16S pro B. vulgatus ATCC 8482 (NCBI GEN ID 5304800). Šedé panely zobrazují variabilní oblasti definované běžně používanými místy pro vázání primerů (doplňková Tabulka 1). Přerušované čáry ukazují očekávaný podíl substitucí nukleotidů, vzhledem k tomu, že v každém genomu je sedm kopií genů 16S., Zdrojem dat jsou poskytovány jako Zdrojový Datový soubor.
s výhodou využít skutečnosti, že Bacteroides vulgatus byl přítomen při vysoké relativní hojnost v našich dvou lidských střevního mikrobiomu vzorků, dále jsme dotázáni, zda intragenomic rozdíly mezi 16 kopií genu by mohly být detekovány in vivo. Zarovnali jsme každou celovečerní sekvenci klasifikovanou jako patřící k našemu Otu B. vulgatus v1–V9 (obr. 3b a doplňující údaje 1) k jediné reprezentativní genové sekvenci B. vulgatus 16S. Následně jsme porovnali výsledné substituční profily nukleotidů (obr., 3c) s profily předpovídanými ze dvou referenčních genomů přítomných v databázi NCBI Refseq19 (obr. 3d).
většina nukleotidové variability přítomen v našem in vivo generované B. vulgatus OTU odráží skutečné rozdíly připadající na intragenomic polymorfismy. Naproti tomu variace pravděpodobně v důsledku chyb sekvenování se objevila nízká a hluboko pod minimální frekvencí ~14%, která by se očekávala, kdyby v každém vzorku byl jediný kmen B.vulgatus se sedmi kopiemi genů 16S v jeho genomu (obr. 3c, přerušované čáry).
i když jsme neznali skutečný počet B., kmeny vulgatus přítomné v každém vzorku in vivo bylo pozoruhodné, že oba substituční profily nukleotidů měly bližší podobnost s kmenem ATCC 8482 než mpk. Variace také existovaly na konkrétních loci, které by mohly potenciálně naznačovat smysluplné rozdíly mezi referenčními genomy in vivo a ATCC 8482. Například v oblasti V5 ATCC 8482 byl zjištěn jediný polymorfismus, který byl přítomen ve třech kopiích 16S (43%). V prvním vzorku in vivo (Scott) byl tento polymorfismus přítomen v 84% čtení, zatímco ve druhém (IronHorse) byl přítomen v 69% čtení., Tato čísla úzce odpovídají číslům očekávaným v případě, že polymorfismus byl přítomen šest a pět ze sedmi genů 16S.
Na závěr jsme se ukázat, že full-délka 16S sekvenování lidského střevního mikrobiomu lze přesně řešit single-nukleotidové substituce, které odrážejí intragenomic rozdíly mezi kopií genu 16S. Přítomnost takové variace naznačuje, že sekvence 16S musí být seskupeny, aby odrážely smysluplné taxonomické jednotky., Pomocí OTUs seskupené na 99% identity, ukazujeme, že full-length 16S má potenciál poskytnout druhy a dokonce i kmen-level taxonomické rozlišení. Analýza mikrobiálních komunit na těchto taxonomických úrovních slibuje poskytnout velmi odlišný pohled na ten, který poskytuje odhady hojnosti na úrovni rodu.
Intragenomic 16 polymorfismů jsou velmi rozšířené
že se prokázalo, že je možné vyřešit intragenomic kopírování varianty in vivo, dále jsme se snažili stanovit, do jaké míry tyto kopie varianty se objevují v taxony běžně vyskytují v lidském střevním mikrobiomem., Dále jsme se snažili zjistit, zda lze tyto profily běžně použít k rozlišení mezi kmeny stejného druhu.
kultivovali jsme 381 taxonů ze střevního mikrobiomu zdravých jedinců zobrazených na obr. 3, stejně jako od jiných osob účastnících se stejné původní studie20 (doplňující údaje 2). My následně provádí full-délka 16S sekvenování genu na izolátů a zarovnané sekvence čte k identifikaci nukleotidové substituce charakteristika intragenomic 16 kopií genu variant.,
Taxonomické zařazení izolátů identifikováno 58 předpokládané druhy (Doplňující Údaje 2), zatímco clustering jeden reprezentativní sekvence pro každý izolát na 99% podobnost vedla v 61 OTUs (s mezi 1 a 73 izolátů přiřazen ke každé OTU). Celkem 349 z 381 sekvenovaných izolátů (54 z 61 OTUs) mělo jeden nebo více SNP, což naznačuje přítomnost 16S genových polymorfismů a 205 jedinečných profilů SNP bylo identifikováno při účtování potenciální chyby sekvenování (obr. 4a a doplňující údaje 2).

Intragenomic 16 genové polymorfismy v lidské střevního mikrobiomu izolátů. umístění SNP přítomných v genech 16S individuálně kultivovaných bakteriálních izolátů. SNP místa byly identifikovány pomocí fázování celovečerních 16S genových sekvencí generovaných pro každý jednotlivý izolát. Osa X označuje polohu podél genu 16S. Osa Y označuje jednotlivé izoláty seskupené na základě jejich odvozené fylogeneze. Tmavě modrá oblast označuje umístění polymorfismu., Pro přehlednost je zobrazeno maximálně pět izolátů patřících ke stejným druhům. Podrobnosti o substitučních profilech nukleotidů pro všechny sekvenované izoláty viz doplňková Data 2. b–d Příklady nukleotidové substituce profily ukazuje napětí-úroveň rozdíly mezi izoláty identifikovány jako patřící do tří bakteriální druhy: b Shigella flexneri; c Bifidobacterium longum; d Collinsella aerofaciens. Pro každý druh jsou zobrazeny dva izolované substituční profily nukleotidů; další příklady však lze nalézt v doplňkových datech 2., Izoláty byly identifikovány jako patřící ke stejnému druhu, pokud byly jejich reprezentativní sekvence přiřazeny ke stejnému OTU při shlukování na 99% identitě sekvence. Taxonomická identifikace byla provedena pomocí BLASTu pro sladění reprezentativních sekvencí s databází NCBI 16S BLAST (viz metody). Šedé panely zobrazují variabilní oblasti definované běžně používanými místy pro vázání primerů (doplňková Tabulka 1). Přerušované čáry ukazují očekávaný podíl substitucí nukleotidů, vzhledem k počtu kopií genů 16S předpovězených pro každý genom., Zdrojem dat jsou poskytovány jako Zdrojový Datový soubor.
Zejména, porovnání SNP profily pro izoláty přiřazeny stejné OTU často vyplynulo, že rozdíly v četnosti Snp, které byly sugestivní rozdíly v intragenomic kopií genu 16S mezi blízce příbuzné taxony. Příklady různých substitučních profilů jsou uvedeny pro tři taxony (obr. 4b-d), které naznačují variaci na úrovni kmene srovnatelnou s variací, kterou jsme v zásadě prokázali Pro E.coli (obr. 2b).,
Na závěr ukázali jsme, že mnoho kultivovat příslušníky lidského střevního mikrobiomu často mají 16 genové polymorfismy, které, pokud jsou správně vyúčtovány, mají potenciál vyřešit kmeny stejného druhu.