Den fulde 16S-genet giver bedre taxonomisk opløsning
~1500 bp 16S rRNA genet består af ni variable regioner veksler hele den meget velbevarede 16S rækkefølge (Fig. 1a). Sekventering af hele genet blev oprindeligt udført ved Sanger sekventering., Dette krævede kloning gener, generering, og samle to til tre læsninger pr klon, og producerer begrænset prøveudtagning dybde på høje omkostninger og indsats. I øjeblikket sekvenserer langt de fleste undersøgelser kun en del af genet, fordi den vidt anvendte Illumina-sekventeringsplatform (højere gennemstrømning, lavere omkostninger, reduceret indsats sammenlignet med Sanger) producerer korte sekvenser (300 300 baser)., Forskellige underregioner af genet er derfor målrettet, lige fra enkelte variable regioner, såsom V4 eller v6, til tre variable regioner, såsom V1–V3 eller V3–V5 (brugt i det humane Mikrobiomprojekt i forbindelse med 454-sekventeringsplatform9).
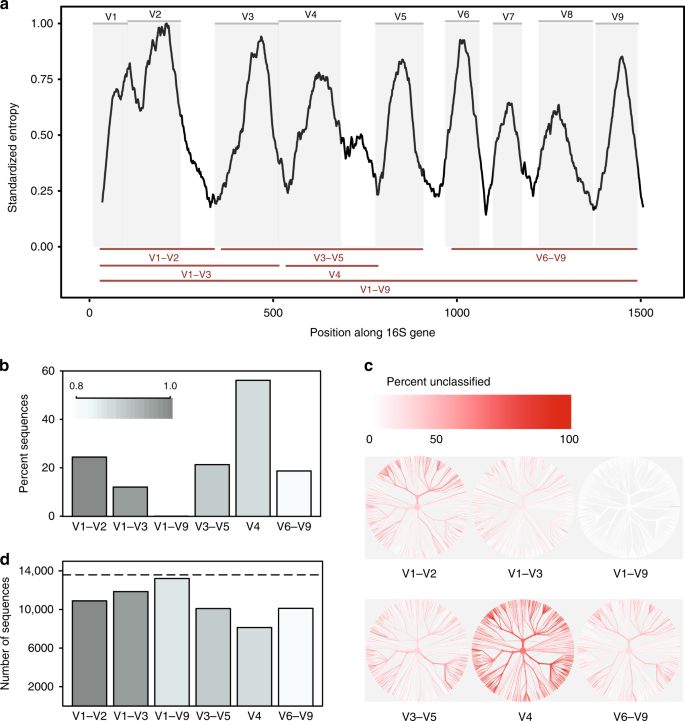
In-silico sammenligning af 16S rRNA variable regioner. en Shannon-entropi på tværs af 16 ‘ erne gen baseret på tilpasningen af en enkelt repræsentativ sekvens for hver kendt art, der findes i Greengens-databasen., Sekvenser blev justeret mod et enkelt reference 16S gen for Escherichia coli K-12 MG1655 (NCBI Gene ID 947777). Grå paneler skildrer variable regioner defineret ved almindeligt anvendte primerbindingssteder (supplerende tabel 1). Variable regioner, der betragtes i denne undersøgelse, vises som røde linjer (nederst). b andel af sekvenser for hver variabel region, der ikke kunne identificeres til artsniveau ved klassificering af hver sekvens i forhold til den referencedatabase, hvorfra den blev afledt ved en konfidenstærskel på 80% (RDP-klassifikator)., C træer baseret på taksonomi af sekvenser til stede i in-silico databasen. Det samme træ er tilvejebragt for hver variabel region. Farven på hver gren afspejler andelen af sekvenser inden for hver clade, der ikke kunne identificeres til artsniveau. d antallet af OTUs oprettet, når clustering sekvenser for hver variabel region på 99% sekvens lighed. Stiplede linje angiver antallet af unikke sekvenser (>1% forskellige) i den oprindelige database., Kildedata leveres som en Kildedatafil
Vi hævder, at målretning af underregioner repræsenterer et historisk kompromis på grund af teknologibegrænsninger 10. I dag er både PacBio-og O .ford Nanopore-sekventeringsplatforme i stand til rutinemæssigt at producere læsninger på over 1500 bp, og højgennemstrømningssekvensering af det fulde 16S-gen bliver stadig mere udbredt., Vi foreslår derfor, at begrundelsen for dette kompromis skal revideres, og vi udførte et simpelt in-silico-eksperiment for at demonstrere fordelen ved fuld længde 16S sekventering i forhold til målretningen af underregioner.
Vi do .nloadede et sæt ikke-redundante (dvs. > 1% forskellige), 16-sekvenser i fuld længde fra en offentlig database (Greengens)., Drage fordel af det faktum, at en betydelig del af disse sekvenser er indarbejdet PCR-primer-bindingssteder, vi klippede dem til at generere in silico-amplifikation for forskellige sub-regioner, som er baseret på placering af PCR-primere, der er almindeligt anvendt i microbiome undersøgelser (Fig. 1a og supplerende tabel 1-2)., Under forudsætning af hver sekvens i vores hentet database repræsenterede en unik arter, vi så brugte en fælles klassificering tilgang (Ribosomets Database Projekt (RDP) classifier11) til at beregne den frekvens, som in-silico-amplifikation for hver sub-regionen kunne give præcise, arter-niveau taksonomisk klassificering (ved hjælp af den oprindelige database som reference). I et andet eksperiment, vi grupperede også vores in-silico ampliconer for at generere OTUs ved forskellige, almindeligt anvendte, sekvenslighedstærskler (97%, 98%, 99%).,
Vi fandt, at underregioner adskiller sig væsentligt i det omfang, de med sikkerhed kunne skelne mellem 16-sekvenserne i fuld længde, der blev brugt til at repræsentere arter (fig. 1b). V4-regionen presterede værst, hvor 56% af ampliconerne i silico ikke med sikkerhed matchede deres oprindelsessekvens på dette taksonomiske niveau. Derimod, naar der blev anvendt en fuld-længde sekvens med alle variable regioner, var det muligt at klassificere næsten alle sekvenser som den korrekte Art (supplerende fig. 1a)., Ændring af databaser og klassificeringskonfidensgrænser påvirkede andelen af in-silico-amplikoner, der kunne matches nøjagtigt, men påvirkede ikke de fremherskende tendenser (supplerende fig. 1a, b).for det andet viste forskellige underregioner bias i den bakterielle ta .a, de var i stand til at identificere (Fig. 1c). For eksempel, V1–V2 region klaret sig dårligt på klassificering af sekvenser, der tilhører phylum Proteobacteria, mens V3–V5-regionen udført dårligt på klassificering af sekvenser, der tilhører phylum Actinobacteria (Supplerende Fig. 2)., Lignende tendenser blev set på slægtsniveau for TA .a af potentiel medicinsk relevans. Selv om den fulde V1–V9-regionen haft de bedste resultater, V6–V9-regionen var især de bedste sub-regionen for klassificering af sekvenser, der tilhører slægterne Clostridium og Staphylococcus V3–V5-regionen har givet gode resultater for Klebsiella, og V1–V3-regionen har givet gode resultater for Escherichia/Shigella (Supplerende Fig. 2 og kildedata).
endelig påvirkede valget af underområde dramatisk antallet af Otus, der blev dannet, når klyngedannelse i silico-amplikoner for at skabe OTUs., Ved klyngedannelse ved 99% sekvensidentitet kunne alle underregioner ikke genskabe antallet af forskellige sekvenser, der var til stede i den oprindelige database; imidlertid udførte v4-regionen igen værst (fig. 1d). Især, det relative antal OTU ‘ er produceret af hver underregion var ikke konsistent ved forskellige identitetstærskler (97%, 98%, 99%, supplerende fig. 3), hvilket indikerer, at opførslen af klyngealgoritmer kan være vanskelig at forudsige, når mængden af information indeholdt i en sekventeret region er meget variabel.,
Som konklusion er målretning af underregioner et historisk kompromis, der var tilstrækkeligt til at identificere ta .aer på slægtsniveau eller derover. Vores enkle in-silico-eksperiment viser imidlertid, at det ikke er gyldigt at antage, at stadig finere klyngning af disse underregioner vil resultere i den forbedrede taksonomiske opløsning, der er nødvendig for at afspejle arter. V1-V3) giver en rimelig tilnærmelse af 16S mangfoldighed, fanger de fleste ikke tilstrækkelig sekvensvariation til at skelne mellem nært beslægtede ta .aer., Vi bemærker også, at diskriminerende polymorfier kan begrænses til specifikke variable regioner; dermed, visse underregioner vil være bedre egnet til at diskriminere nært beslægtede medlemmer af visse ta .aer.
16S genkopi-varianter afspejler variation i belastningsniveau
klyngedannelse af 16S-sekvenser i OTUs har historisk tjent to formål. Først, det har fjernet mindre artifactual sekvens varianter på grund af PCR amplifikation og sekventering fejl, når kollapse sekvenser i grupper. Anden, Det har kollapset legitime sekvens varianter, der findes mellem nært beslægtede bakterielle ta .a., Selvom sidstnævnte måske ikke altid er ønskelig, det er grunden til, at du ikke kan skelne mellem bakterieta .a, hvis 16S-sekvenser varierer med en hastighed, der er lavere end den fejl, der opstår på en bestemt sekventeringsplatform.
for nylig har fremskridt i CCS dramatisk forbedret fejlfrekvenser på langlæste sekventeringsplatforme. På samme tid, beregningsmetoder har gjort det muligt at skelne mellem legitim vs artifactual sekvens variation., Disse teknologiske og metodologiske fremskridt betyder, at forskere nu har potentialet til at udføre sekventering med høj gennemstrømning, der nøjagtigt kan detektere enkelt-nukleotidvarianter på tværs af hele 16S-genet.
Selv om det er fristende at antage, at single-nucleotide varianter kan repræsentere forskellige, nært beslægtede taxa, vi advare mod denne alt for forsimplet fortolkning på grund af det faktum, at mange bakterielle genomer indeholder flere polymorfe kopier af 16S gene12,13,14., Vi udførte PacBio CCS sekventering af en 36 arter bakteriel mock samfund (supplerende tabel 3 og supplerende Fig. 4) for at demonstrere (i) at 16S-sekvensen for mange bakterier varierer mellem operoner inden for det samme genom, og (ii) at højgennemstrømningssekvensering er tilstrækkelig nøjagtig til at løse disse intragenomiske forskelle.
vi justerede PacBio 16S-sekvenser i fuld længde til en referencedatabase, der indeholder en enkelt repræsentativ 16S-sekvens for hvert medlem af vores mock-samfund og brugte justeringsstatistikken til at evaluere nøjagtigheden af denne sekventeringsmetode., At sammenligne antallet af overkørsler, der anvendes til at generere et CCS med forekomst af enkelt-nukleotid substitutioner, insertions og udeladelser er angivet, at ti passerer kunne minimere disse kombinerede fejl til et minimum frekvens af < 1.0% (selv om det var bemærkelsesværdigt, at den mindste opnåelige fejl varierede mellem sekventering kører Supplerende Fig. 5). Vi observerede dog et tilfælde af sletningsfejl med placeringen homopolymer kører i vores referencesekvenser (supplerende Fig., 6), som ikke var nukleotidspecifik og blev forværret af længden af den sekventerede homopolymer (supplerende fig. 7). Vi er efterfølgende valideret sletninger i Escherichia coli 16S-genet ved hjælp af Illumina hele genomet shotgun (WGS) sekventering, som viste, at kun én af sletninger forekommer i PacBio sekvenser var ægte (Supplerende Fig. 8).,
det Godtgjort, at CCS-sekventering kan producere 16S læser med en lav frekvens af substitution fejl, vi næste begrundet, at en del af substitution fejl inden præcist afstemt læser bør afspejle variationen kan henføres til 16S polymorfier inden for en art’ genome12. Læser for eksempel justeret til E. coli-stammen K-12 substr. MG1655 viste en substitutionsprofil, som afspejlede nøjagtigt det forudsagt ved at tilpasse alle syv af 16 ‘ erne sekvenser, der vides at være til stede i dette genome15 (fig. 2a, C)., Vi var yderligere i stand til at validere støkiometrien af disse nukleotidsubstitutioner ved at kvantificere variation i sammenligneligt justeret Illumina .gs læser (fig. 2b) og demonstrere, at en lignende substitutionsprofil var reproducerbar på tværs af flere sekventeringskørsler (supplerende fig. 9)., Justeringer til andre referencesekvenser i vores mock-samfund viste en lignende tendens med rigelige substitutioner lokaliseret til specifikke basepositioner langs 16S-genet, selvom vi bemærker, at signal-til-støj-forholdet steg markant, når det pågældende 16S-gen havde færre end 100 justerede læsninger (supplerende fig. 10).

Polymorfier i E. coli-16S rRNA-sekvenser af gener. a positionen og hyppigheden af substitutioner, der optræder i E., coli stamme K-12 mg1655 v1-v9 ampliconer genereret fra vores mock samfund og sekventeret på PacBio RS II platform. B positionen og hyppigheden af substitutioner i læsninger genereret fra genomisk sekventering af den isolerede E. coli-stamme K-12 MG1655 på Illumina mise. – platformen. Forstørrede regioner viser respektive positioner i justeringen af alle syv 16S-gener, der er til stede i E. coli K-12 mg1655-referencegenomet. 16 ‘ erne sekvens fra rrnd operon (**) bruges som reference for alle SNP udfasning. c den forudsagte nukleotidsubstitutionsprofil for E., coli K-12 MG1655 baseret på justering af de syv 16S gensekvenser, der er til stede i referencegenomet. D den forudsagte substitutionsprofil for E. coli O157 Sakai baseret på justering af de syv 16S gensekvenser, der er til stede i referencegenomet. Grå paneler skildrer variable regioner defineret ved almindeligt anvendte primerbindingssteder (supplerende tabel 1). Stiplede linjer angiver den forventede andel af nukleotidsubstitutioner, da der er syv 16S genkopier inden for hvert genom., Kildedata leveres som en Kildedatafil
observationen af, at langvarig sekventering kan identificere 16S polymorfier inden for det samme genom, har vigtige konsekvenser. For det første demonstrerer det,at det ikke er gyldigt at antage, at high-throughput sekvens læser forskellige med en eller få nukleotider repræsenterer en særskilt ta .a6, 16. Inden for et enkelt genom kan to eller flere 16-sekvenser være identiske, mens andre kan være unikke., Tilsvarende kan nogle homologe 16S loci beholde identisk sekvens mellem to nært beslægtede stammer, mens andre kan have divergeret i en eller få nukleotidpositioner. I denne sammenhæng, alle eu-plan eller taksonomiske fortolkning af 16S data bør ideelt set tage højde for, at den relative tæthed af 16S-sekvenser, der hidrører fra den meget nært beslægtede taxa, der vil afspejle en lineær kombination af (i) den frekvens, som hver unik sekvens er repræsenteret i hele genomer og (ii) den relative tæthed af genomer for hver taxon.,
for det andet, selvom intragenomisk 16S-sekvensvariation komplicerer analyse på samfundsniveau, har den også potentialet til at øge kraften i 16S-genet til at diskriminere mellem nært beslægtede ta .a, fordi det muliggør sekvensbaseret sammenligning at strække sig over flere divergerende loci. For eksempel, er tilstrækkelig nukleotid variation findes for at skelne E. coli stamme K-12 MG1655 fra enterohemorrhagic stamme O157 Sakai (Fig. 2c, d)., Dermed, Vi argumenterer for, at, når det er passende redegjort for, flere polymorfe 16S-kopier er ikke en ulempe, der skal overses, snarere vil de gøre det muligt for 16S-genet at blive brugt i mikrobiomanalyse på stammeniveau. Vi bemærker også, at kraften i intragenomic 16S sekvens variation at diskriminere nært beslægtede ta .a er tilbøjelige til at aftage, når delvis 16S sekvenser anvendes. For eksempel SNPs skelne E. coli stammer K-12 MG1655 (Fig. 2c) fra O157 Sakai (Fig. 2d) findes i variable regioner V1, V2, V6 og v9.,
16S-polymorfier kan løses in vivo
Mikrobiomsamfund er ofte komplekse, der findes i forskellige biokemiske miljøer (f.afføring, spyt, sputum osv.) og indeholder mange hundrede unikke ta .a, hvis relative overflod spænder over et bredt dynamisk område. Denne kompleksitet er ikke godt repræsenteret i hverken in-silico eller mock samfund eksperimenter. Vi udførte derfor et yderligere eksperiment for at demonstrere, at sekventering af det fulde 16S-gen, mens vi tegner os for intragenomiske 16S SNP ‘ er, kan løse nært beslægtede bakterieta .a in vivo.,
Vi udførte PacBio CCS–sekventering af V1-v9-regionen for fire humane afføringsprøver indsamlet fra raske voksne frivillige. Til sammenligning, vi har sekventeret V1–V3-regionen ved hjælp af Illumina MiSeq, og for at give et benchmark for arter-niveau taksonomiske kvantificering, vi udførte metagenomic WGS (mWGS) sekventering ved hjælp af Illumina NextSeq. For at evaluere, i hvilket omfang hver af disse sekventeringsmetoder kan løse nært beslægtede ta .aer, fokuserede vi på slægten Bacteroides., Ud over at være rigelig i den menneskelige tarm, er denne slægt meget forskelligartet, der indeholder flere arter, der kan udøve både gode og dårlige virkninger på menneskers sundhed17. Det har også været anvendt tidligere som en model ta .on til at demonstrere nytten af 16S genet til høj opløsning taksonomisk analyse18.
Når vi beregnet Bacteroides overflod på Slægt niveau, v1–v9 sekventering og V1–V3 sekventering produceret sammenlignelige resultater., Begge fremgangsmåder identificerede to personer med lav Bacteroides relativ overflod (~10-25%) og to personer med høj Bacteroides relativ overflod (~40-60%; Fig. 3a). Kvantificering af artsniveau via M .gs-sekventering afslørede imidlertid langt større mangfoldighed, med forskellige Bacteroides-arter dominerende i tarmen hos hver enkelt person (fig. 3b og supplerende Data 1). Når klyngedannelse OTUs ved 99% identitet, både v1–V9 og V1–V3 sekventering var i stand til at afspejle denne art-niveau variation (fig., 3b), med den bemærkelsesværdige undtagelse, at V1–V3-sekventering ikke registrerede Bacteroides intestinalis, hvilket var rigeligt i en af de fire humane tarmmikrobiomprøver. Baseret på disse resultater konkluderer vi, at når de anvendes sammen med en passende identitetstærskel (f.99%), har Otu-baserede tilgange potentialet til at løse artsdiversitet observeret i den menneskelige tarm. Vi bemærker endvidere, at selvom 16-sekventering i fuld længde kan være optimal til analyse af arter, kan meget informative variable regioner (f.V1-V3) også være tilstrækkelige til dette formål.,

påvisning af bakteroider i humane afføringsprøver. a den relative forekomst af slægten Bacteroides i fire humane afføringsprøver kvantificeret ved anvendelse af enten v1-v9 ampliconer (axis–akse) eller V1-V3 ampliconer (y-akse). b den relative forekomst af Bacteroides arter i de samme fire prøver. Arter overflod blev kvantificeret fra m .gs sekventering eller fra V1–V3/v1–v9 OTUs genereret ved 99% identitet., Overflod er vist for de mest udbredte arter som kvantificeret ved m .gs (for overflod estimater af alle Bacteroides arter påvist af hver platform, se supplerende tabel 5). C Nukleotidsubstitutionsprofiler genereret ved at tilpasse alle v1–v9 ampliconsekvenser tildelt den enkelte OTU identificeret som Bacteroides vulgatus. Profiler er vist for de to afføringsprøver med høj B. vulgatus relativ overflod (IronHorse og Scott). D nukleotidsubstitutionsprofiler forudsagt fra referencegenomerne af to forskellige B. vulgatus-stammer ATCC 848239 og mpk40., I både c og d blev nukleotidsubstitutioner identificeret i forhold til et enkelt reference 16S-gen for B. vulgatus ATCC 8482 (NCBI Gene ID 5304800). Grå paneler skildrer variable regioner defineret ved almindeligt anvendte primerbindingssteder (supplerende tabel 1). Stiplede linjer angiver den forventede andel af nukleotidsubstitutioner, da der er syv 16S genkopier inden for hvert genom., Kilde data, som kan bruges som en Kilde Data-fil
drage fordel af det faktum, at Bacteroides vulgatus var til stede ved høj relativ tæthed i to af vores menneskelige gut microbiome prøver, næste gang vi bliver spurgt, om intragenomic variation mellem 16S gen kopier kunne påvises i vivo. Vi justerede hver fuldlængdesekvens klassificeret som tilhørende vores B. vulgatus V1-V9 OTUs (Fig. 3b og supplerende Data 1) til en enkelt repræsentativ B. vulgatus 16S gensekvens. Vi sammenlignede derefter de resulterende nukleotidsubstitutionsprofiler (fig., 3c) med profiler forudsagt fra to referencegenomer til stede i NCBI Refse.database19 (fig. 3d).
størstedelen af nukleotidvariationen til stede i vores in vivo genererede B. vulgatus OTU afspejlede sand variation, der kan henføres til intragenomiske polymorfier. I modsætning hertil variation sandsynligvis på grund af sekventering fejl optrådte lave og langt under det minimale ~14% frekvens, der kunne forventes, hvis der var en enkelt B. vulgatus stamme i hver prøve med syv 16S gen-kopier i dens genom (Fig. 3c, stiplede linjer).
selvom vi ikke vidste det sande antal B., vulgatus-stammer til stede i hver In vivo-prøve, det var bemærkelsesværdigt, at begge nukleotidsubstitutionsprofiler havde tættere lighed med stamme ATCC 8482 end MPK. Variation eksisterede også ved specifikke loci, der potentielt kunne indikere meningsfulde forskelle mellem in vivo og ATCC 8482 referencegenomer. For eksempel blev en enkelt polymorfisme påvist i v5-regionen i ATCC 8482, som var til stede i tre 16S-kopier (43%). I den første in vivo-prøve (Scott) var denne polymorfisme til stede i 84% af læsningerne, mens den i den anden (IronHorse) var til stede i 69% af læsningerne., Disse tal svarer nøje til de forventede tal, hvis en polymorfisme var til stede seks og fem ud af syv 16S gener, henholdsvis.
afslutningsvis viser vi, at 16-sekventering i fuld længde af det humane tarmmikrobiom nøjagtigt kan løse enkelt-nukleotidsubstitutioner, der afspejler intragenom variation mellem 16-genkopier. Tilstedeværelsen af en sådan variation indikerer, at 16S-sekvenser skal grupperes for at afspejle meningsfulde taksonomiske enheder., Ved hjælp af OTUs grupperet med 99% identitet viser vi, at 16 ‘ er i fuld længde har potentialet til at levere arter og endda stamme-niveau taksonomisk opløsning. Analyse af mikrobielle samfund på disse taksonomiske niveauer lover at give et meget andet perspektiv end det, der ydes af genus-niveau overflod estimater.
Intragenomic 16S polymorfier er meget udbredt
Der har vist, at det er muligt at løse intragenomic kopi varianter in vivo, har vi søgt at fastslå, i hvilket omfang en sådan kopi varianter vises i taxa, der almindeligvis findes i den menneskelige tarm microbiome., Vi søgte endvidere at fastslå, om saadanne profiler rutinemæssigt kan bruges til at skelne mellem stammer af samme art.
Vi dyrkede 381 ta .a fra tarmmikrobiomet hos de sunde individer afbildet i Fig. 3, samt fra andre personer, der deltager i samme oprindelige undersøgelse20 (supplerende Data 2). Vi udførte efterfølgende fuld længde 16S gensekvensering på isolater og justerede sekventerede læsninger for at identificere nukleotidsubstitutioner, der er karakteristiske for intragenomiske 16S genkopieringsvarianter.,
Taksonomisk klassificering af isolater, der er identificeret 58 formodede arter (Supplerende Data, 2), mens clustering en enkelt repræsentant rækkefølge for hver isolere på 99% lighed resulterede i 61 OTUs (med mellem 1 og 73 isolater, der er tildelt hver OTU). I alt 349 381 sekventeret isolater (54 61 OTUs) havde en eller flere SNP, der angiver tilstedeværelsen af 16S-genet polymorfier, og 205 unikke SNP profiler blev identificeret, når der tegner sig for potentielle sekventering fejl (Fig. 4a og supplerende Data 2).

Intragenomic 16S-genet polymorfier i menneskets tarmsystem microbiome isolater. en placering af SNP ‘er til stede i 16’ erne gener af individuelt dyrkede bakterieisolater. SNP steder blev identificeret gennem udfasning fuld længde 16S gensekvenser genereret for hver enkelt isolat. X-aksen angiver position langs 16S genet. Y-aksen betegner individuelle isolater grupperet baseret på deres udledte fylogeni. Mørkeblå region angiver placeringen af en polymorfisme., For klarhedens skyld vises maksimalt fem isolater, der tilhører den samme art. For nærmere oplysninger om nukleotidsubstitutionsprofiler for alle sekventerede isolater, se supplerende Data 2. b–d Eksempler på nukleotid substitution profiler, der viser, stamme-niveau forskelle mellem isolater, der er identificeret som tilhørende tre bakterielle arter: b Shigella flexneri; c Bifidobacterium longum; d Collinsella aerofaciens. For hver art er der vist to isolatnukleotidsubstitutionsprofiler; yderligere eksempler kan dog findes i supplerende Data 2., Isolater blev identificeret som tilhørende den samme art, hvis deres repræsentative sekvenser blev tildelt den samme OTU, når klyngedannelse ved 99% sekvensidentitet. Taksonomisk identifikation blev udført ved hjælp af BLAST at tilpasse repræsentative sekvenser til NCBI 16S BLAST database (Se metoder). Grå paneler skildrer variable regioner defineret ved almindeligt anvendte primerbindingssteder (supplerende tabel 1). Stiplede linjer angiver den forventede andel af nukleotidsubstitutioner i betragtning af antallet af 16S genkopier forudsagt for hvert genom., Kilde data, som kan bruges som en Kilde Data-fil
Især, at sammenligne SNP profiler for isolater, der er tildelt til den samme OTU ofte afsløret forskelle i hyppigheden af SNPs, der blev orienteret af forskelle i intragenomic 16S gen kopier mellem nært beslægtede taxa. Eksempler på forskellige substitutionsprofiler er vist for tre ta .aer (fig. 4b-d), som tyder på variation i stamme-niveau, der kan sammenlignes med den, vi i princippet demonstrerede for E. coli (Fig . 2b).,
afslutningsvis viser vi, at mange af de kulturelle medlemmer af det humane tarmmikrobiom ofte besidder 16S-genpolymorfismer, som, når de er korrekt redegjort for, har potentialet til at løse stammer af samme art.