RNA-Sequenzierung (Wang 2009) ersetzt in vielen Labors schnell Genexpressionsmikroarrays. Mit RNA-seq können Sie RNAs quantifizieren, entdecken und profilieren. Für diese Technik werden mRNA (und andere RNAs) zuerst in cDNA umgewandelt. Die cDNA wird dann als Eingabe für eine Sequenzierungsbibliotheksvorbereitung der nächsten Generation verwendet. In diesem Artikel werde ich einen kurzen Überblick über RNA-seq geben und die wichtigsten Methoden vorstellen, die heute verwendet werden.
Warum ist RNA-Seq „besser“ als Mikroarrays?,
RNA-seq hat mehrere Vorteile gegenüber Mikroarrays:
Mit RNA-seq können Sie mehr als nur differentielle Genexpression abfragen. Obwohl Mikroarrays für Exon-Level-und microRNA-Analysen verfügbar sind, sind die meisten Benutzer immer noch an einer grundlegenden, wahrscheinlich 3′ voreingenommenen, differentiellen Genexpression interessiert. Mit RNA-seq können Sie sich die Kodierung und Nicht-Kodierung von RNA, die Spleiß-und allelspezifische Expression und möglicherweise bald cDNA-Sequenzen in voller Länge ansehen, sodass keine Isoformen abgeleitet oder zusammengesetzt werden müssen.,
Mikroarrays sind ebenfalls voreingenommen, da wir entscheiden müssen, welchen Inhalt wir auf dem Array platzieren möchten. Da RNA-seq keine Sonden oder Primer verwendet, leiden die Daten unter viel geringeren Verzerrungen (obwohl ich nicht sagen möchte, dass RNA-seq keine hat).
RNA-seq liefert digitale Daten in Form von ausgerichteten Lesezahlen, was zu einem sehr breiten Dynamikbereich führt und die Erkennungsempfindlichkeit für seltene Transkripte verbessert.
Es ist auch sehr kostengünstig für Mikroarrays, da heute zwischen 6-30 Proben in einer einzigen Illumina-Sequenzierungsspur multiplext werden können.,
Schließlich können Sie einen RNA-seq-Datensatz neu analysieren, sobald weitere Informationen zum Transkriptom verfügbar sind. Wenn ein Papier veröffentlicht wird, das eine interessante Spleißvariante in einem ähnlichen System wie das zeigt, an dem Sie arbeiten, möchten Sie vielleicht zurückgehen und sich dieses Spleißen in Ihren Beispielen ansehen.und du hättest bereits die Daten dazu.
Wie funktioniert RNA-Seq?
Es gibt viele Methoden zur Durchführung eines RNA-seq-Experiments. Tatsächlich entwickeln sich die Techniken so schnell, dass es schwierig sein kann, zu entscheiden, welche verwendet werden soll., Eine grundlegende Entscheidung ist zwischen 1) random-primed cDNA-Synthese aus double-stranded cDNA-oder 2 -) – RNA-ligation-Methoden (geprüft und verglichen, Levin 2010). Die meisten Menschen verwenden die erste Methode und müssen dann eine weitere Wahl zwischen einem strangspezifischen Protokoll und einem Protokoll treffen, das dies nicht ist. Die Methode, die am häufigsten in meinem Labor verwendet wird, ist Illuminas TruSeq RNA-seq, ein zufällig grundiertes cDNA-Synthese-nicht-strangspezifisches Protokoll.
Sobald Sie eine Sequenzierungsbibliothek haben, wird sie auf eine bestimmte Tiefe sequenziert, die davon abhängt, was Sie mit den Daten tun möchten., Diese Lesevorgänge sind auf das Genom oder Transkriptom ausgerichtet und werden gezählt, um die differentielle Genexpression zu bestimmen, oder weiter analysiert, um die Spleiß-und Isoformexpression zu bestimmen. Die meisten Menschen sequenzieren RNA mit gepaarten End-50-100bp-Methoden. Die Ausnahme ist die MikroRNA-Sequenzierung, da dies in den meisten Fällen nur eine Single-End – 36bp-Sequenzierung erfordert.
Unsere RNA-Seq-Methode
Wir verwenden zwischen 100 ng und 1 µg Gesamt-RNA als Eingang zu einem mRNA-Capture mit oligo-dT beschichteten Magnetperlen. Die mRNA wird fragmentiert und dann wird eine zufällig grundierte cDNA-Synthese durchgeführt., Die resultierende Doppelstrang-cDNA wird als Eingang zu einer Standard-Illumina-Bibliotheksvorbereitung verwendet, die Endreparatur, Adapterligation und PCR-Verstärkung umfasst, um Ihnen eine Bibliothek zur Sequenzierung zur Verfügung zu stellen.
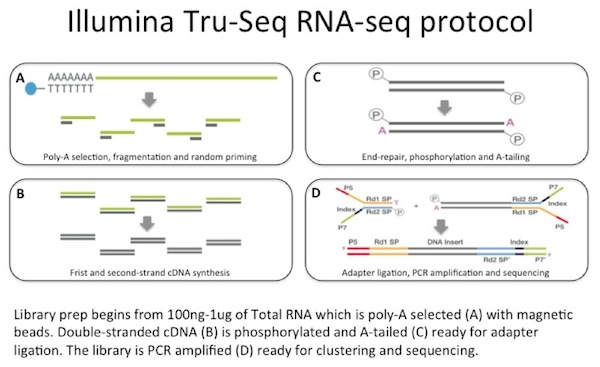
Warum sollten Sie Sich Mit Diesen Informationen Beschäftigen?
Es wurde eine große Diskussion über die anti-sense-Transkription und Ihre biologische Relevanz. Wenn Sie an einer einfachen differentiellen Genexpression interessiert sind, werden Stranginformationen Ihrem Experiment nicht viel hinzufügen, aber Ihr Protokoll komplexer machen., Trotzdem können Sie die am weitesten verbreitete Methode ohne zu viel zusätzlichen Aufwand ausführen. Verwenden Sie dazu während der cDNA-Synthese des 2. Strangs Uracil zur Inkorporation anstelle von Thymin. Folgen Sie der Illumina Library Prep wie gewohnt, aber nach der Adapterligatur und vor der PCR-Amplifikation Uracil-DNA-Glykosylase hinzufügen, um den 2ndstrand abzubauen. Dies führt dazu, dass alle Lesevorgänge in derselben Ausrichtung beginnen, sodass Sie bestimmen können, welcher Strang in Ihrem Sample transkribiert wurde.
Was kann man eigentlich mit RNA-Seq machen?,
RNA-seq ist ein leistungsstarkes und vielseitiges Tool, das in den letzten Jahren weit verbreitet war. Ich habe einige meiner Favoriten ausgewählt (einige von der Arbeit in der von mir verwalteten Kerneinrichtung), um zu veranschaulichen, was Sie mit der RNA-Sequenzierung tun können.
- Jabbari, et al. verwendete RNA-seq, um Psoriasis zu untersuchen und neue Gene für die funktionelle Analyse zu finden. Sie verglichen ihre RNA-seq-Daten mit veröffentlichten Array-Studien und fanden 1700 neue Kandidaten. Diese wurden durch qPCR validiert, und der Vergleich mit funktionellen Datenbanken für Psoriasis unterstützte ihre Rolle in der Pathogenese.
- Kutter, et al., verwendete RNA-seq in einer Studie zur Erhaltung der RNA-Polymerase-III-Bindung bei Säugetieren zur Validierung der Expression von Genen, die von Pol III besetzt sind, wie von ChIP-seq untersucht.
- Mercer, et al. kombinierte RNA-seq-und Microarray-basierte Erfassung zur Identifizierung und Charakterisierung seltener Transkripte, die normalerweise nicht nachweisbar sind. Die gezielten Transkripte erhöhten sich im Sequenzleseüberfluss von 0,21% vor der Erfassung auf 80% nach der Erfassung., Sie fanden mehr als 200 zuvor nicht annotierte Isoformen für fast 50 proteinkodierende Loci, einschließlich einer neuen alternativen Isoform von TP53, einem sehr gut charakterisierten Gen. Dies deutet darauf hin, dass das Genom und das Transkriptom noch viel komplexer zu lösen sind.
Zusammenfassend ist RNA-seq immer noch ein sich entwickelndes Werkzeug, ist aber in den meisten Fällen Mikroarrays vorzuziehen. Es ist empfindlicher, robuster und kann kostengünstiger sein. Welche Projekte planen Sie jetzt für Ihr Projekt?,
Jabbari et al: transkriptionsanalyse von Psoriasis Mithilfe von RNA-seq Enthüllt, die Zuvor nicht identifizierten Differentiell Exprimierten Gene. Zeitschrift für investigative Dermatologie 2011.
– Kutter et al: Pol III-Bindung in sechs Säugetieren zeigt unter Naturschutz Aminosäure isotypes trotz der Unterschiede zwischen den tRNA-Genen. Nature Genetics 2011.
Levin et al: Umfassende vergleichende Analyse von strand-specific RNA-Sequenzierung Methoden. Natur Methoden 2010.
Mercer et al: Gezielte RNA-Sequenzierung zeigt die Tiefe Komplexität des menschlichen Transkriptom. Nature Biotechnology 2012.,
Wang et al: RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009.
Ursprünglich am 25. Mai 2012 veröffentlicht. Aktualisiert und überarbeitet am August 16, 2015.

Hat das Ihnen geholfen? Dann teilen Sie bitte mit Ihrem Netzwerk.