el gen 16S completo proporciona una mejor resolución Taxonómica
el gen 16S rRNA ~1500 bp comprende nueve regiones variables intercaladas a lo largo de la secuencia 16S altamente conservada (Fig. 1a). Secuenciación el gen entero fue logrado originalmente por Sanger secuenciación., Esto requirió clonar genes, generar y ensamblar dos o tres lecturas por clon, y producir una profundidad de muestreo limitada a un alto costo y esfuerzo. Actualmente, sin embargo, la gran mayoría de los estudios secuencian solo una parte del gen, porque la plataforma de secuenciación Illumina ampliamente utilizada (mayor rendimiento, menor costo, menor esfuerzo en comparación con Sanger) produce secuencias cortas ( ≤ 300 bases)., Por lo tanto, se dirigen diferentes subregiones del gen, que van desde regiones variables únicas, como V4 o V6, hasta tres regiones variables, como V1-V3 o V3–V5 (utilizadas en el proyecto de microbioma humano junto con la plataforma de secuenciación 4549).
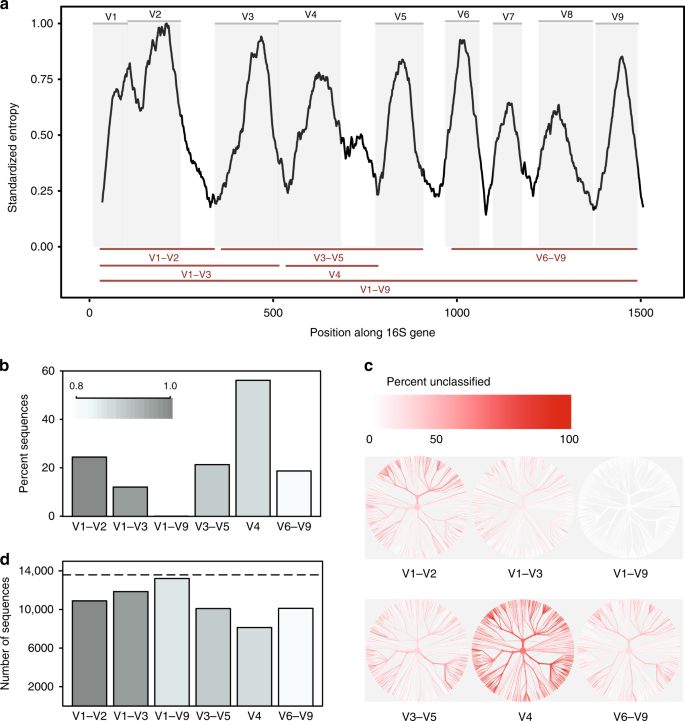
comparación In-silico de regiones variables rRNA de 16S. una entropía de Shannon a través del gen 16S basada en la alineación de una sola secuencia representativa para cada especie conocida presente en la base de datos de Greengenes., Las secuencias se alinearon contra un único gen de referencia 16S para Escherichia coli K-12 MG1655 (NCBI Gene ID 947777). Los paneles grises representan regiones variables definidas por los sitios de unión de imprimación comúnmente utilizados (tabla suplementaria 1). Las regiones variables consideradas en este estudio se muestran como líneas rojas (abajo). B proporción de secuencias para cada región variable que no se pudo identificar a nivel de especie al clasificar cada secuencia en la base de datos de referencia de la que se derivó a un umbral de confianza del 80% (clasificador RDP)., árboles c basados en la taxonomía de secuencias presentes en la base de datos in-silico. El mismo árbol se proporciona para cada región variable. El color de cada rama refleja la proporción de secuencias dentro de cada clado que no pudieron ser identificadas a nivel de especie. d el número de OTUs creados al agrupar secuencias para cada región variable con una similitud de secuencia del 99%. La línea discontinua indica el número de secuencias únicas (> 1% diferente) en la base de datos original., Los datos de origen se proporcionan como un archivo de datos de origen
argumentamos que la segmentación de subregiones representa un compromiso histórico, debido a las restricciones tecnológicas10. Hoy en día, las plataformas de secuenciación de nanoporos PacBio y Oxford son capaces de producir lecturas rutinarias superiores a 1500 bp y la secuenciación de alto rendimiento del gen 16S completo es cada vez más frecuente., Por lo tanto, sugieren que la justificación para este compromiso debe ser revisada y se realizó un sencillo de in-silico experimento para demostrar la ventaja de longitud completa la secuenciación de 16S sobre la orientación de las sub-regiones.
descargamos un conjunto de secuencias 16S no redundantes (es decir, > 1% diferentes) de una base de Datos pública (Greengenes)., Aprovechando el hecho de que una proporción sustancial de estas secuencias incorporaba sitios de unión de cebadores de PCR, los recortamos para generar amplicones in-silico para diferentes subregiones, basados en la ubicación de cebadores de PCR comúnmente utilizados en estudios de microbioma (Fig. 1a y cuadros complementarios 1 y 2)., Asumiendo que cada secuencia en nuestra base de datos descargada representaba una especie única, luego utilizamos un enfoque de clasificación común (el proyecto de base de datos de ribosomas (RDP) classifier11) para calcular la frecuencia con la que los amplicones in-silico para cada subregión podrían proporcionar una clasificación taxonómica precisa a nivel de especie (utilizando la base de datos original como referencia). En un segundo experimento, también agrupamos nuestros amplicones in-silico para generar OTUs en diferentes umbrales de similitud de secuencias comúnmente utilizados(97%, 98%, 99%).,
encontramos que las subregiones diferían sustancialmente en la medida en que podían discriminar con confianza entre las secuencias 16S de longitud completa utilizadas para representar especies (Fig. 1b). La región V4 tuvo el peor desempeño, con el 56% de los amplicones in-silico fallando en igualar con confianza su secuencia de origen en este nivel taxonómico. Por el contrario, cuando se utilizó una secuencia de longitud completa con todas las regiones variables, fue posible clasificar casi todas las secuencias como la especie correcta (Fig. 1a)., La alteración de las bases de datos y los umbrales de confianza de la clasificación afectaron la proporción de amplicones in-silico que podían ser emparejados con precisión, pero no influyeron en las tendencias prevalecientes (Fig.suplementaria. 1a, b).
en segundo lugar, diferentes subregiones mostraron sesgo en los taxones bacterianos que fueron capaces de identificar (Fig. 1c). Por ejemplo, la región V1–V2 tuvo un desempeño pobre en la clasificación de secuencias pertenecientes al filo Proteobacteria, mientras que la región V3–V5 tuvo un desempeño pobre en la clasificación de secuencias pertenecientes al filo Actinobacteria (suplemento Fig. 2)., Se observaron tendencias similares a nivel de Género para taxones de posible relevancia médica. Aunque la región v1–V9 completa produjo consistentemente los mejores resultados, la región V6–V9 fue notablemente la mejor subregión para clasificar secuencias pertenecientes a los géneros Clostridium y Staphylococcus, la región V3–V5 produjo buenos resultados para Klebsiella, y la región V1–V3 produjo buenos resultados para Escherichia/Shigella (suplemento Fig. 2 y datos de origen).
finalmente, la elección de la subregión afectó dramáticamente el número de OTUs formados al agruparse en amplicones in-silico para crear Otus., Al agruparse con una identidad de secuencia del 99%, todas las subregiones no pudieron recrear el número de secuencias distintas presentes en la base de datos original; sin embargo, la región V4 nuevamente tuvo el peor desempeño (Fig. 1d). En particular, el número relativo de Uto producidas por cada subregión no fue consistente en diferentes umbrales de identidad(97%, 98%, 99%, suplemento Fig. 3), lo que indica que el comportamiento de los Algoritmos de agrupamiento puede ser difícil de predecir cuando la cantidad de información contenida dentro de una región secuenciada es altamente variable.,
en conclusión, la selección de subregiones representa un compromiso histórico que fue suficiente para la identificación de taxones a nivel de género o superior. Sin embargo, nuestro simple experimento in-silico demuestra que no es válido asumir que una agrupación cada vez más fina de estas subregiones resultará en la resolución Taxonómica mejorada necesaria para reflejar las especies. Aunque algunas subregiones (por ejemplo, V1–V3) proporcionan una aproximación razonable de la diversidad de 16S, la mayoría no capturan suficiente variación de secuencia para discriminar entre taxones estrechamente relacionados., También observamos que los polimorfismos discriminantes pueden restringirse a regiones variables específicas; por lo tanto, ciertas subregiones serán más adecuadas para discriminar miembros estrechamente relacionados de ciertos taxones.
las variantes de copia del gen 16S reflejan la variación a nivel de cepa
la agrupación de secuencias 16S en OTUs ha servido históricamente para dos propósitos. En primer lugar, ha eliminado variantes menores de secuencia artifactual debido a la amplificación de PCR y errores de secuenciación al colapsar secuencias en grupos. En segundo lugar, ha colapsado Variantes de secuencia legítimas que existen entre taxones bacterianos estrechamente relacionados., Aunque esto último puede no ser siempre deseable, es lógico que no se pueda distinguir entre los taxones bacterianos cuyas secuencias de 16S varían a una velocidad que es menor que el error encontrado en una plataforma de secuenciación en particular.
recientemente, los avances en CCS han mejorado drásticamente las tasas de error de las plataformas de secuenciación de larga lectura. Al mismo tiempo, los métodos computacionales han hecho posible distinguir entre la variación legítima frente a la variación de secuencia artificial., Estos avances tecnológicos y metodológicos significan que los investigadores ahora tienen el potencial de realizar una secuenciación de alto rendimiento que puede detectar con precisión Variantes de un solo nucleótido en todo el gen 16S.
aunque es tentador asumir que las variantes de un solo nucleótido pueden representar taxones distintos y estrechamente relacionados, advertimos contra esta interpretación excesivamente simplista debido al hecho de que muchos genomas bacterianos contienen múltiples copias polimórficas del gen 16S12,13,14., Se realizó la secuenciación PacBio CCS de una comunidad simulada bacteriana de 36 especies(tabla suplementaria 3 y Fig. suplementaria. 4) demostrar (i) que la secuencia 16S de muchas bacterias varía entre operones dentro del mismo genoma y (ii) que la secuenciación de alto rendimiento es lo suficientemente precisa para resolver estas diferencias intragenómicas.
alineamos las secuencias 16s de PacBio a una base de datos de referencia que contiene una única secuencia 16S representativa para cada miembro de nuestra comunidad simulada y utilizamos las estadísticas de alineación para evaluar la precisión de este enfoque de secuenciación., Comparando el número de pasadas utilizadas para generar un CCS con la ocurrencia de sustituciones, inserciones y deleciones de un solo nucleótido, se indicó que diez pasadas podrían minimizar estos errores combinados a una frecuencia mínima de < 1.0% (aunque fue notable que el error mínimo alcanzable varió entre corridas de secuenciación; suplementario Fig. 5). Sin embargo, sí observamos una coincidencia de errores de eliminación con las corridas de homopolímero de ubicación en nuestras secuencias de referencia (Fig., 6), que no era específico de nucleótidos y se vio exacerbado por la longitud del homopolímero secuenciado (suplemento Fig. 7). Posteriormente validamos deleciones dentro del gen Escherichia coli 16S utilizando la secuenciación Illumina whole genome shotgun (WGS), lo que demostró que solo una de las deleciones ocurridas en las secuencias de PacBio era genuina (suplemento Fig. 8).,
satisfechos de que la secuenciación CCS puede producir lecturas de 16S con una baja frecuencia de errores de sustitución, razonamos a continuación que una proporción de los errores de sustitución dentro de lecturas alineadas con precisión debería reflejar la variación atribuible a polimorfismos de 16S dentro del genoma de una especie12. Por ejemplo, lee alineado a la cepa K-12 de E. coli substr. MG1655 mostró un perfil de sustitución, que reflejaba exactamente lo predicho al alinear las siete secuencias 16S conocidas por estar presentes en este genomo15 (Fig. 2a, c)., Además, pudimos validar la estequiometría de estas sustituciones de nucleótidos cuantificando la variación en lecturas de Illumina WGS comparativamente alineadas (Fig. 2b)y demostrar que un perfil de sustitución similar era reproducible a través de múltiples secuencias de secuenciación (Fig. 9)., Los alineamientos con otras secuencias de referencia en nuestra comunidad simulada mostraron una tendencia similar de sustituciones abundantes localizadas a posiciones de base específicas a lo largo del gen 16S, aunque observamos que la relación señal-ruido aumentó significativamente cuando el gen 16S en cuestión tenía menos de 100 lecturas alineadas (Fig.suplementaria. 10).

polimorfismos en secuencias del gen rRNA de E. coli 16S. a la posición y frecuencia de las sustituciones que aparecen en E., coli cepa K-12 MG1655 v1-V9 amplicones generados a partir de nuestra comunidad simulada y secuenciados en la plataforma PacBio RS II. b la posición y frecuencia de las sustituciones en las lecturas generadas a partir de la secuenciación genómica de la cepa aislada de E. coli K-12 MG1655 en la plataforma Illumina MiSeq. Las regiones magnificadas muestran posiciones respectivas en la alineación de los siete genes 16S presentes en el genoma de referencia de E. coli K-12 MG1655. La secuencia 16S del operón rrnD ( * * ) se utiliza como referencia para todas las fases SNP. C el perfil de sustitución de nucleótidos previsto de E., coli K-12 MG1655 basado en la alineación de las siete secuencias del gen 16S presentes en el genoma de referencia. d el perfil de sustitución predicho de E. coli O157 Sakai basado en la alineación de las siete secuencias del gen 16S presentes en el genoma de referencia. Los paneles grises representan regiones variables definidas por los sitios de unión de imprimación comúnmente utilizados (tabla suplementaria 1). Las líneas discontinuas indican la proporción esperada de sustituciones de nucleótidos, dado que hay siete copias de genes 16S dentro de cada genoma., Los datos de origen se proporcionan como un archivo de datos de origen
la observación de que la secuenciación de larga lectura puede identificar polimorfismos 16S dentro del mismo genoma tiene implicaciones importantes. En primer lugar,demuestra que no es válido asumir que las lecturas de secuencias de alto rendimiento que difieren en uno o pocos nucleótidos representan un taxa distinto6, 16. Dentro de un solo genoma, dos o más secuencias 16S pueden ser idénticas, mientras que otras pueden ser únicas., En consecuencia, algunos loci 16S homólogos pueden retener la secuencia idéntica entre dos cepas estrechamente relacionadas, mientras que otros pueden haber divergido en una o pocas posiciones de nucleótidos. En este contexto, cualquier interpretación a nivel comunitario o taxonómico de los datos del 16S debería tener en cuenta idealmente el hecho de que la abundancia relativa de las secuencias del 16S derivadas de taxones muy estrechamente relacionados reflejará una combinación lineal de (i) la frecuencia con la que cada secuencia única se representa a través de los genomas y (ii) la abundancia relativa de los genomas para cada taxón.,
en segundo lugar, aunque la variación intragenómica de la secuencia 16S complica el análisis a nivel comunitario, también tiene el potencial de aumentar el poder del gen 16S para discriminar entre taxones estrechamente relacionados, porque permite la comparación basada en secuencias para extenderse a través de múltiples loci divergentes. Por ejemplo, existe suficiente variación de nucleótidos para distinguir la cepa K-12 MG1655 de E. coli de la cepa enterohemorrágica O157 Sakai (Fig. 2c, d)., Por lo tanto, argumentamos que, cuando se contabilizan adecuadamente, las copias polimórficas múltiples de 16S no son un inconveniente que se debe pasar por alto, sino que permitirán que el gen 16S se use en el análisis del microbioma a nivel de cepa. También observamos que el poder de la variación intragenómica de la secuencia 16S para discriminar taxones estrechamente relacionados es probable que disminuya cuando se utilizan secuencias parciales de 16S. Por ejemplo, los SNP que distinguen las cepas de E. coli K-12 MG1655 (Fig. 2c) de O157 Sakai (Fig. 2d) se encuentran en las regiones variables V1, V2, V6 y V9.,
los polimorfismos 16S se pueden resolver in vivo
Las comunidades del microbioma son a menudo complejas, existiendo en diversos ambientes bioquímicos (por ejemplo, heces, saliva, esputo, etc.).) y que contiene muchos cientos de taxones únicos cuya abundancia relativa abarca un amplio rango dinámico. Esta complejidad no está bien representada en experimentos comunitarios in-silico o simulados. Por lo tanto, realizamos un experimento adicional para demostrar que la secuenciación del gen 16S completo mientras se tiene en cuenta el SNPs intragenómico 16S puede resolver taxones bacterianos estrechamente relacionados in vivo.,
se realizó la secuenciación PacBio CCS de la región V1–V9 para cuatro muestras de heces humanas colectadas de voluntarios adultos sanos. Para la comparación, secuenciamos la región V1-V3 utilizando el Illumina MiSeq y, para proporcionar un punto de referencia para la cuantificación Taxonómica a nivel de especie, realizamos la secuenciación metagenómica WGS (mWGS) utilizando el Illumina NextSeq. Para evaluar la medida en que cada uno de estos enfoques de secuenciación puede resolver taxones estrechamente relacionados, Nos centramos en el género Bacteroides., Además de ser abundante en el intestino humano, este género es muy diverso, conteniendo múltiples especies que pueden ejercer efectos tanto buenos como malos sobre la salud humana17. También se ha utilizado previamente como un taxón modelo para demostrar la utilidad del gen 16S para el análisis taxonómico de alta resolución18.
Cuando calculamos la abundancia de Bacteroides a nivel de género, la secuenciación V1–V9 y la secuenciación V1–V3 produjeron resultados comparables., Ambos abordajes identificaron dos individuos con baja abundancia relativa de Bacteroides (~10-25%) y dos individuos con alta abundancia relativa de Bacteroides (~40-60%; Fig. 3a). Sin embargo, la cuantificación a nivel de especie a través de la secuenciación de mWGS reveló una diversidad mucho mayor, con una especie de Bacteroides diferente dominante en el intestino de cada individuo (Fig. 3b y datos complementarios 1). Al agrupar OTUs con una identidad del 99%, tanto la secuenciación V1–V9 como la V1–V3 fueron capaces de reflejar esta variación a nivel de especie (Fig., 3b), con la notable excepción de que la secuenciación V1–V3 no detectó Bacteroides intestinalis, que era abundante en una de las cuatro muestras de microbioma intestinal humano. Sobre la base de estos resultados, concluimos que, cuando se utilizan junto con un umbral de identidad apropiado (por ejemplo, 99%), los enfoques basados en OTU tienen el potencial de resolver la diversidad a nivel de especie observada en el intestino humano. Observamos además que, aunque la secuenciación completa de 16S puede ser óptima para el análisis a nivel de especies, las regiones variables altamente informativas (por ejemplo, V1-V3) también pueden ser adecuadas para este propósito.,

la Detección de Bacteroides en muestras de heces humanas. a la abundancia relativa del género Bacteroides en cuatro muestras de heces humanas cuantificadas usando amplicones V1-V9 (eje x) o amplicones V1-V3 (eje y). B La abundancia relativa de especies Bacteroides en las mismas cuatro muestras. La abundancia de especies se cuantificó a partir de la secuenciación de mWGS o de Uto V1–V3/V1-V9 generadas al 99% de identidad., La abundancia se muestra para las especies más abundantes cuantificada por mWGS (para las estimaciones de abundancia de todas las especies de Bacteroides detectadas por cada plataforma, ver la tabla suplementaria 5). C perfiles de sustitución de nucleótidos generados alineando todas las secuencias de amplicón v1-V9 asignadas a la única OTU identificada como Bacteroides vulgatus. Se muestran los perfiles de las dos muestras de heces con abundancia relativa alta de B. vulgatus (IronHorse y Scott). D Nucleotide substitution profiles predicted from the reference genomes of two different B. vulgatus strains ATCC 848239 and mpk40., Tanto en c como en d, se identificaron sustituciones de nucleótidos en relación con un único gen de referencia 16S para B. vulgatus ATCC 8482 (NCBI Gene ID 5304800). Los paneles grises representan regiones variables definidas por los sitios de unión de imprimación comúnmente utilizados (tabla suplementaria 1). Las líneas discontinuas indican la proporción esperada de sustituciones de nucleótidos, dado que hay siete copias de genes 16S dentro de cada genoma., Los datos de origen se proporcionan como un archivo de datos de origen
aprovechando el hecho de que Bacteroides vulgatus estaba presente en alta abundancia relativa en dos de nuestras muestras de microbioma intestinal humano, a continuación preguntamos si la variación intragenómica entre copias del gen 16S podía detectarse in vivo. Alineamos cada secuencia completa clasificada como perteneciente a nuestro OTUs B. vulgatus v1-V9 (Fig. 3b y datos suplementarios 1) a una única secuencia representativa del gen B. vulgatus 16S. Luego comparamos los perfiles de sustitución de nucleótidos resultantes (Fig., 3c) con perfiles predichos a partir de dos genomas de referencia presentes en la base de datos NCBI Refseq19 (Fig. 3d).
la mayor parte de la variación de nucleótidos presente en nuestro B. VULGATUS OTU generado in vivo reflejó una variación verdadera atribuible a polimorfismos intragenómicos. En contraste, la variación probablemente debida a errores de secuenciación parecía baja y muy por debajo de la frecuencia mínima de ~14% que se esperaría si hubiera una sola cepa de B. vulgatus en cada muestra con siete copias del gen 16S en su genoma (Fig. 3C, líneas discontinuas).
aunque no sabíamos el número verdadero de B., cepas de vulgatus presentes en cada muestra in vivo, fue notable que ambos perfiles de sustitución de nucleótidos tenían un parecido más cercano a la cepa ATCC 8482 que a la mpk. También existían variaciones en loci específicos que podrían indicar diferencias significativas entre los genomas de Referencia in vivo y ATCC 8482. Por ejemplo, se detectó un único polimorfismo en la región V5 de ATCC 8482, que estaba presente en tres copias de 16S (43%). En la primera muestra in vivo (Scott) este polimorfismo estuvo presente en el 84% de las lecturas, mientras que en la segunda (IronHorse) estuvo presente en el 69% de las lecturas., Estos números corresponden estrechamente a los números esperados si un polimorfismo estuviera presente seis y cinco de los siete genes 16S, respectivamente.
en conclusión, mostramos que la secuenciación completa de 16S del microbioma intestinal humano puede resolver con precisión las sustituciones de un solo nucleótido que reflejan la variación intragenómica entre las copias génicas de 16S. La presencia de tal variación indica que las secuencias 16S deben agruparse para reflejar unidades taxonómicas significativas., Utilizando Otus agrupados en un 99% de identidad, mostramos que los 16 de longitud completa tienen el potencial de proporcionar una resolución Taxonómica a nivel de especie e incluso de cepa. El análisis de las comunidades microbianas en estos niveles taxonómicos promete proporcionar una perspectiva muy diferente a la proporcionada por las estimaciones de Abundancia a nivel de género.
los polimorfismos Intragenómicos 16S son altamente prevalentes
habiendo demostrado que es posible resolver Variantes de copia intragenómicas in vivo, a continuación buscamos establecer el grado en que tales variantes de copia aparecen en taxones comúnmente encontrados dentro del microbioma intestinal humano., Además, tratamos de establecer si tales perfiles se pueden utilizar rutinariamente para distinguir entre cepas de la misma especie.
se cultivaron 381 taxones del microbioma intestinal de los individuos sanos representados en la Fig. 3, así como de otras personas participantes en el mismo estudio original20 (datos complementarios 2). Posteriormente realizamos secuenciación completa del gen 16S en aislados y lecturas secuenciadas alineadas para identificar sustituciones de nucleótidos características de variantes intragenómicas de copia del gen 16S.,
la clasificación taxonómica de aislados identificó 58 especies putativas (datos suplementarios 2), mientras que la agrupación de una sola secuencia representativa para cada aislado con una similitud del 99% resultó en 61 Uto (con entre 1 y 73 aislados asignados a cada UTO). En total, 349 de 381 aislados secuenciados (54 de 61 Uto) tenían uno o más SNP, lo que indica la presencia de polimorfismos del gen 16S, y se identificaron 205 perfiles únicos de SNP al considerar un posible error de secuenciación (Fig. 4a y datos complementarios 2).

polimorfismos del gen Intragenómico 16S en aislados de microbioma intestinal humano. localización de SNPs presentes en los genes 16S de aislados bacterianos cultivados individualmente. Las localizaciones de SNP fueron identificadas a través del phasing de secuencias gene 16S de longitud completa generadas para cada aislado individual. El eje X denota la posición a lo largo del gen 16S. El eje y denota aislados individuales agrupados en base a su filogenia inferida. Azul oscuro indica la ubicación de un polimorfismo., Para mayor claridad, se muestran un máximo de cinco cepas pertenecientes a la misma especie. Para más detalles sobre los perfiles de sustitución de nucleótidos para todos los aislados secuenciados, ver datos suplementarios 2. b-D ejemplos de perfiles de sustitución de nucleótidos que muestran diferencias a nivel de cepa entre aislados identificados como pertenecientes a tres especies bacterianas: B Shigella flexneri; C Bifidobacterium longum; D Collinsella aerofaciens. Para cada especie, se muestran dos perfiles de sustitución de nucleótidos aislados; sin embargo, se pueden encontrar ejemplos adicionales en datos suplementarios 2., Los aislados se identificaron como pertenecientes a la misma especie si sus secuencias representativas se asignaron a la misma OTU cuando se agruparon en una identidad de secuencia del 99%. La identificación taxonómica se realizó mediante BLAST para alinear secuencias representativas con la base de datos NCBI 16S BLAST (ver Métodos). Los paneles grises representan regiones variables definidas por los sitios de unión de imprimación comúnmente utilizados (tabla suplementaria 1). Las líneas discontinuas indican la proporción esperada de sustituciones de nucleótidos, dado el número de copias genéticas de 16S predichas para cada genoma., Los datos de origen se proporcionan como un archivo de datos de origen
notablemente, la comparación de perfiles de SNP para aislados asignados a la misma OTU reveló con frecuencia diferencias en la frecuencia de SNPs que sugirieron diferencias en las copias del gen intragenómico 16S entre taxones estrechamente relacionados. Se muestran ejemplos de diferentes perfiles de sustitución para tres taxones (Fig. 4b-d), que sugieren una variación del nivel de deformación comparable a la que demostramos en principio para E. coli (Fig. 2b).,
en conclusión, mostramos que muchos de los miembros cultivables del microbioma intestinal humano con frecuencia poseen polimorfismos del gen 16S, que, cuando se contabilizan adecuadamente, tienen el potencial de resolver cepas de la misma especie.