le gène 16S complet offre une meilleure résolution taxonomique
le gène de l’ARNr 16S ~1500 PB comprend neuf régions variables intercalées tout au long de la séquence 16S hautement conservée (Fig. 1a). Séquençage le gène entier a été accompli à l’origine par séquençage Sanger., Cela nécessitait de cloner des gènes, de générer et d’assembler deux à trois lectures par clone, et de produire une profondeur d’échantillonnage limitée à un coût et à un effort élevés. Actuellement, cependant, la grande majorité des études ne séquencent qu’une partie du gène, car la plate-forme de séquençage Illumina largement utilisée (débit plus élevé, coût réduit, effort réduit par rapport à Sanger) produit des séquences courtes ( ≤ 300 bases)., Différentes sous-régions du gène sont donc ciblées, allant de régions variables uniques, telles que V4 ou V6, à trois régions variables, telles que V1–V3 ou V3–V5 (utilisées dans le projet Microbiome humain en conjonction avec la plateforme de séquençage 4549).
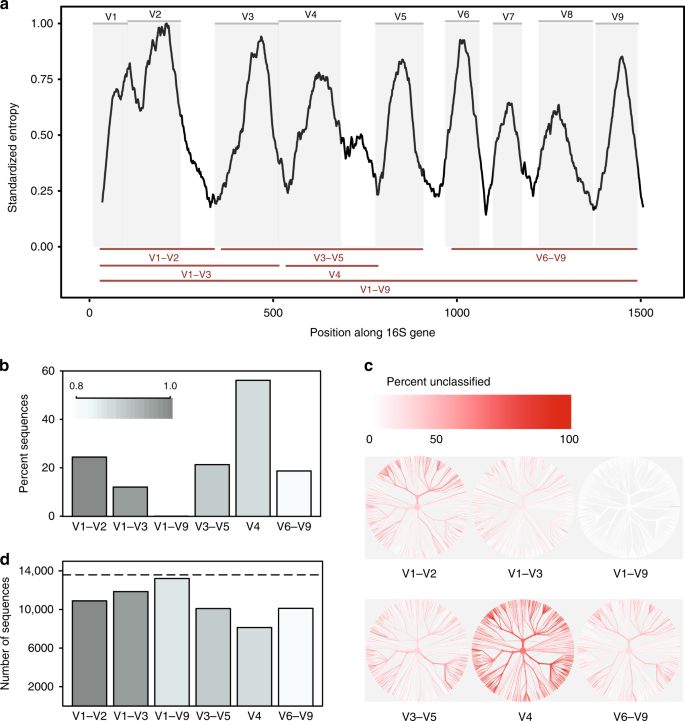
comparaison in-silico des régions variables de l’ARNr 16S. une entropie de Shannon à travers le gène 16S basée sur l’alignement d’une seule séquence représentative pour chaque espèce connue présente dans la base de données Greengenes., Les séquences ont été alignées sur un seul gène de référence 16S pour Escherichia coli K-12 MG1655 (gène NCBI ID 947777). Les panneaux gris représentent des régions variables définies par les sites de liaison d’amorce couramment utilisés (tableau supplémentaire 1). Les régions variables considérées dans cette étude sont indiquées sous forme de lignes rouges (en bas). B Proportion de séquences pour chaque région variable qui n’ont pas pu être identifiées au niveau de l’espèce lors de la classification de chaque séquence par rapport à la base de données de référence à partir de laquelle elle a été dérivée à un seuil de confiance de 80% (Classificateur RDP)., C arbres basés sur la taxonomie des séquences présentes dans la base de données in-silico. Le même arbre est fourni pour chaque région variable. La couleur de chaque branche reflète la proportion de séquences au sein de chaque clade qui n’ont pas pu être identifiées au niveau de l’espèce. D le nombre D’Otu créées lors du regroupement de séquences pour chaque région variable à 99% de similarité de séquence. La ligne pointillée indique le nombre de séquences uniques (>1% différent) dans la base de données d’origine., Les données Source sont fournies sous forme de fichier de données Source
Nous soutenons que le ciblage des sous-régions représente un compromis historique, en raison des restrictions technologiques 10. Aujourd’hui, les plates-formes de séquençage PacBio et Oxford Nanopore sont capables de produire régulièrement des lectures supérieures à 1500 bp et le séquençage à haut débit du gène 16S complet devient de plus en plus répandu., Nous suggérons donc que la justification de ce compromis doit être réexaminée et nous avons réalisé une expérience in silico simple pour démontrer l’avantage du séquençage 16S complet par rapport au ciblage des sous-régions.
nous avons téléchargé un ensemble de séquences 16S non redondantes (c’est-à-dire> 1% différentes), de pleine longueur à partir d’une base de données publique (Greengenes)., Profitant du fait qu’une proportion importante de ces séquences incorporait des sites de liaison des amorces de PCR, nous les avons rognés pour générer des amplicons in-silico pour différentes sous-régions, en fonction de l’emplacement des amorces de PCR couramment utilisées dans les études sur le microbiome (Fig. 1a et tableaux supplémentaires 1 à 2)., En supposant que chaque séquence de notre base de données téléchargée représentait une espèce unique, nous avons ensuite utilisé une approche de classification commune (le classificateur RDP (Ribosome Database Project) 11) pour calculer la fréquence à laquelle les amplicons in-silico pour chaque sous-région pourraient fournir une classification taxonomique précise au niveau des espèces (en utilisant la base de données originale comme référence). Dans une deuxième expérience, nous avons également regroupé nos amplicons in-silico pour générer des Otu à différents seuils de similarité de séquence couramment utilisés(97%, 98%, 99%).,
Nous avons constaté que les sous-régions différaient considérablement dans la mesure où elles pouvaient discriminer en toute confiance les séquences 16S sur toute la longueur utilisées pour représenter les espèces (Fig. 1b). La région V4 a été la moins performante, 56% des amplicons in-silico ne parvenant pas à correspondre avec confiance à leur séquence d’origine à ce niveau taxonomique. En revanche, lorsqu’une séquence pleine longueur avec toutes les régions variables a été utilisée, il a été possible de classer presque toutes les séquences comme l’espèce correcte (fig. supplémentaire. 1a)., La modification des bases de données et des seuils de confiance de classification a eu une incidence sur la proportion d’amplicons in-silico qui pouvaient être appariés avec précision, mais n’a pas influencé les tendances dominantes (fig. supplémentaire. 1a, b).
Deuxièmement, différentes sous-régions ont montré un biais dans les taxons bactériens qu’elles ont pu identifier (Fig. 1c). Par exemple, la région V1–V2 s’est mal comportée lors de la classification des séquences appartenant au phylum Proteobacteria, tandis que la région V3–V5 s’est mal comportée lors de la classification des séquences appartenant au phylum Actinobacteria (fig. 2)., Des tendances similaires ont été observées au niveau du genre pour les taxons présentant un intérêt médical potentiel. Bien que la région v1-V9 complète ait toujours produit les meilleurs résultats, la région V6–V9 était notamment la meilleure sous-région pour classer les séquences appartenant aux genres Clostridium et Staphylococcus, la région V3–V5 a produit de bons résultats pour Klebsiella et la région V1–V3 a produit de bons résultats pour Escherichia/Shigella (fig. 2 et données sources).
enfin, le choix de la sous-région a considérablement affecté le nombre d’Uto formés lors du regroupement d’amplicons in-silico pour créer des Uto., Lors du clustering à 99% d’identité de séquence, toutes les sous-régions n’ont pas réussi à recréer le nombre de séquences distinctes présentes dans la base de données d’origine; cependant, la région V4 a de nouveau été la moins performante (Fig. 1d). Notamment, le nombre relatif D’Uto produites par chaque sous-région n’était pas cohérent à différents seuils d’identité(97%, 98%, 99%, Fig.supplémentaire 3), indiquant que le comportement des algorithmes de clustering peut être difficile à prévoir lorsque la quantité d’informations contenues dans une région séquencée est très variable.,
En conclusion, le ciblage des sous-régions représente un compromis historique qui était suffisant pour l’identification des taxons au niveau du genre ou au-dessus. Cependant, notre simple Expérience in-silico démontre qu’il n’est pas valide de supposer que le regroupement toujours plus fin de ces sous-régions entraînera l’amélioration de la résolution taxonomique nécessaire pour refléter les espèces. Bien que certaines sous-régions (p. ex., V1–V3) fournissent une approximation raisonnable de la diversité 16S, la plupart ne capturent pas suffisamment de variation de séquence pour faire la distinction entre des taxons étroitement apparentés., Nous notons également que les polymorphismes discriminants peuvent être limités à des régions variables spécifiques; ainsi, certaines sous-régions seront mieux adaptées pour discriminer les membres étroitement apparentés de certains taxons.
les variantes de copie du gène 16S reflètent la variation au niveau de la souche
Le Regroupement des séquences 16S en OTUs a historiquement servi à deux fins. Tout d’abord, il a supprimé les variantes de séquence artefactuelles mineures en raison de l’amplification par PCR et des erreurs de séquençage lors de la réduction des séquences en groupes. Deuxièmement, il a réduit les variantes de séquence légitimes qui existent entre des taxons bactériens étroitement apparentés., Bien que ce dernier ne soit pas toujours souhaitable, il va de soi que vous ne pouvez pas distinguer entre les taxons bactériens dont les séquences 16S varient à un taux inférieur à l’erreur rencontrée sur une plate-forme de séquençage particulière.
récemment, les progrès de la SCC ont considérablement amélioré les taux d’erreur des plates-formes de séquençage à lecture longue. Dans le même temps, les méthodes de calcul ont permis de distinguer entre la variation légitime et la variation de séquence artifactuelle., Ces avancées technologiques et méthodologiques signifient que les chercheurs ont maintenant le potentiel d’effectuer un séquençage à haut débit capable de détecter avec précision des variantes à un seul nucléotide sur l’ensemble du gène 16S.
bien qu’il soit tentant de supposer que les variants à un seul nucléotide peuvent représenter des taxons distincts et étroitement apparentés, nous mettons en garde contre cette interprétation trop simpliste en raison du fait que de nombreux génomes bactériens contiennent de multiples copies polymorphes du gène 16S12, 13,14., Nous avons effectué le séquençage PacBio CCS d’une communauté simulée bactérienne de 36 Espèces (tableau supplémentaire 3 et figure supplémentaire. 4) démontrer (i) que la séquence 16S de nombreuses bactéries varie entre les opérons du même génome et (ii) que le séquençage à haut débit est suffisamment précis pour résoudre ces différences intragénomiques.
Nous avons aligné des séquences 16s pleine longueur PacBio sur une base de données de référence contenant une seule séquence 16S représentative pour chaque membre de notre communauté simulée et nous avons utilisé les statistiques d’alignement pour évaluer la précision de cette approche de séquençage., La comparaison du nombre de passages utilisés pour générer un SCC avec l’apparition de substitutions, d’insertions et de délétions d’un seul nucléotide a indiqué que dix passages pouvaient réduire ces erreurs combinées à une fréquence minimale de < 1,0% (bien qu’il soit notable que l’erreur minimale réalisable variait entre les cycles de séquençage; 5). Cependant, nous avons observé une coïncidence d’erreurs de délétion avec l’emplacement des homopolymères dans nos séquences de référence (fig. supplémentaire., 6), qui n’était pas spécifique aux nucléotides et était exacerbée par la longueur de l’homopolymère séquencé (fig. 7). Nous avons ensuite validé les délétions dans le gène Escherichia coli 16S en utilisant le séquençage Illumina whole genome shotgun (WGS), ce qui a démontré qu’une seule des délétions survenant dans les séquences PacBio était authentique (fig. supplémentaire. 8).,
convaincus que le séquençage CCS peut produire des lectures 16S avec une faible fréquence d’erreurs de substitution, nous avons ensuite estimé qu’une proportion des erreurs de substitution dans les lectures correctement alignées devrait refléter la variation attribuable aux polymorphismes 16S dans le génome d’une espèce12. Par exemple, les lectures sont alignées sur la souche K-12 de E. coli. MG1655 a montré un profil de substitution, qui reflétait exactement celui prédit en alignant les sept séquences 16S connues pour être présentes dans ce génome15 (Fig. 2 bis, c)., Nous avons également pu valider la stœchiométrie de ces substitutions nucléotidiques en quantifiant la variation des lectures WGS Illumina alignées de manière comparable (Fig. 2b) et démontrer qu’un profil de substitution similaire était reproductible sur plusieurs séries de séquençage (fig. supplémentaire. 9)., Les alignements avec d’autres séquences de référence dans notre communauté simulée ont montré une tendance similaire de substitutions abondantes localisées à des positions de base spécifiques le long du gène 16S, bien que nous remarquions que le rapport signal sur bruit augmentait significativement lorsque le gène 16S en question avait moins de 100 lectures alignées (fig. supplémentaire. 10).

polymorphismes dans les séquences du gène de l’ARNr 16S D’E. coli. a la position et la fréquence des substitutions apparaissant dans E., coli souche K–12 MG1655 v1-V9 amplicons générés à partir de notre communauté simulée et séquencés sur la plate-forme PacBio RS II. b La position et la fréquence des substitutions dans les lectures générées par le séquençage génomique de la souche isolée D’E. coli K-12 MG1655 sur la plateforme Illumina MiSeq. Les régions agrandies montrent des positions respectives dans l’alignement des sept gènes 16S présents dans le génome de référence D’E. coli K-12 MG1655. La séquence 16S de l’opéron rrnD ( * * ) est utilisée comme référence pour tous les phasages SNP. c le profil prédit de substitution nucléotidique de E., coli K-12 MG1655 basé sur l’alignement des sept séquences du gène 16S présentes dans le génome de référence. D le profil de substitution prévu d’E. coli O157 Sakai basé sur l’alignement des sept séquences du gène 16S présentes dans le génome de référence. Les panneaux gris représentent des régions variables définies par les sites de liaison d’amorce couramment utilisés (tableau supplémentaire 1). Les lignes pointillées indiquent la proportion attendue de substitutions nucléotidiques, étant donné qu’il existe sept copies du gène 16S dans chaque génome., Les données sources sont fournies sous forme de fichier de données Source
l’observation selon laquelle le séquençage à lecture longue peut identifier des polymorphismes 16S dans le même génome a des implications importantes. Tout d’abord,il démontre qu’il n’est pas valide de supposer que les lectures de séquences à haut débit différant par un ou quelques nucléotides représentent un taxa distinct 6, 16. Au sein d’un même génome, deux séquences 16S ou plus peuvent être identiques, tandis que d’autres peuvent être uniques., En conséquence, certains locus homologues 16S peuvent conserver une séquence identique entre deux souches étroitement apparentées, tandis que d’autres peuvent avoir divergé à une ou quelques positions nucléotidiques. Dans ce contexte, toute interprétation communautaire ou taxonomique des données 16S devrait idéalement tenir compte du fait que l’abondance relative des séquences 16S provenant de taxons très étroitement apparentés reflétera une combinaison linéaire de (i) la fréquence avec laquelle chaque séquence unique est représentée dans les génomes et (ii) l’abondance relative des génomes pour chaque taxon.,
deuxièmement, bien que la variation intragénomique de la séquence 16S complique l’analyse au niveau de la communauté, elle a également le potentiel d’augmenter le pouvoir du gène 16S de discriminer entre des taxons étroitement apparentés, car elle permet la comparaison basée sur la séquence pour s’étendre à plusieurs loci divergents. Par exemple, il existe une variation nucléotidique suffisante pour distinguer la souche K-12 MG1655 D’E. coli de la souche entérohémorragique O157 Sakai (fig. 2c, d)., Ainsi, nous soutenons que, lorsqu’elles sont dûment prises en compte, les multiples copies polymorphes 16S ne sont pas un inconvénient à négliger, elles permettront plutôt d’utiliser le gène 16s dans l’analyse du microbiome au niveau de la souche. Nous notons également que le pouvoir de la variation intragénomique de la séquence 16S pour discriminer des taxons étroitement apparentés est susceptible de diminuer lorsque des séquences partielles 16S sont utilisées. Par exemple, SNPs distinguant les souches D’E. coli K-12 MG1655 (Fig. 2c) de O157 Sakai (fig. 2d) se trouvent dans les régions variables V1, V2, V6 et V9.,
les polymorphismes 16S peuvent être résolus in vivo
les communautés du Microbiome sont souvent complexes, existant dans divers environnements biochimiques (p. ex., selles, salive, expectorations, etc.) et contenant plusieurs centaines de taxons uniques dont l’abondance relative couvre une large plage dynamique. Cette complexité n’est pas bien représentée dans les expériences communautaires in-silico ou simulées. Nous avons donc effectué une expérience supplémentaire pour démontrer que le séquençage du gène 16S complet tout en tenant compte des SNP 16S intragénomiques peut résoudre des taxons bactériens étroitement apparentés in vivo.,
Nous avons effectué le séquençage PacBio CCS de la région V1–V9 pour quatre échantillons de selles humaines prélevés sur des volontaires adultes en bonne santé. À titre de comparaison, nous avons séquencé la région V1–V3 à l’aide du MiSeq Illumina et, pour fournir une référence pour la quantification taxonomique au niveau des espèces, nous avons effectué un séquençage métagénomique WGS (mWGS) à l’aide du NextSeq Illumina. Pour évaluer dans quelle mesure chacune de ces approches de séquençage peut résoudre des taxons étroitement liés, nous nous sommes concentrés sur le genre Bacteroides., En plus d’être abondant dans l’intestin humain, ce genre est très diversifié, contenant de multiples espèces qui peuvent avoir des effets à la fois bons et mauvais sur la santé humaine17. Il a également été utilisé précédemment comme taxon modèle pour démontrer l’utilité du gène 16S pour l’analyse taxonomique à haute résolution 18.
lorsque nous avons calculé L’abondance des Bacteroides au niveau du genre, le séquençage V1–V9 et le séquençage V1–V3 ont produit des résultats comparables., Les deux approches ont identifié deux individus avec une faible abondance relative de Bacteroides (~10-25%) et deux individus avec une abondance relative de Bacteroides élevée (~40-60%; Fig. 3a). Cependant, la quantification au niveau des espèces par séquençage mWGS a révélé une bien plus grande diversité, avec une espèce Bacteroides différente dominante dans l’intestin de chaque individu (Fig. 3b et données supplémentaires 1). Lors du regroupement des OTUs à 99% d’identité, le séquençage V1–V9 et V1–V3 a pu refléter cette variation au niveau de l’espèce (Fig., 3b), à l’exception notable que le séquençage V1–V3 n’a pas détecté Bacteroides intestinalis, qui était abondant dans l’un des quatre échantillons de microbiome intestinal humain. Sur la base de ces résultats, nous concluons que, lorsqu’elles sont utilisées conjointement avec un seuil d’identité approprié (p. ex., 99%), les approches basées sur L’OTU ont le potentiel de résoudre la diversité au niveau des espèces observée dans l’intestin humain. Nous notons en outre que, bien que le séquençage complet du 16S puisse être optimal pour l’analyse au niveau de l’espèce, des régions variables très informatives (p. ex., V1-V3) peuvent également être adéquates à cette fin.,

la Détection de Bacteroides de l’homme dans les échantillons de selles. a l’abondance relative du genre Bacteroides dans quatre échantillons de selles humaines quantifiée à l’aide d’amplicons V1–V9 (axe des abscisses) ou V1-V3 (axe des ordonnées). b l’abondance relative des espèces de Bacteroides dans les mêmes quatre échantillons. L’abondance des espèces a été quantifiée à partir du séquençage de mWGS ou à partir D’OTUs V1–V3/V1–V9 générés à 99% d’identité., L’abondance est indiquée pour les espèces les plus abondantes telles que quantifiées par les MWg (pour les estimations de l’abondance de toutes les espèces de Bacteroides détectées par chaque plate-forme, voir le tableau supplémentaire 5). profils de substitution de nucléotides c générés en alignant toutes les séquences d’amplicon V1–V9 assignées à L’OTU unique identifiée comme Bacteroides vulgatus. Des profils sont présentés pour les deux échantillons de selles présentant une abondance relative élevée de B. vulgatus (IronHorse et Scott). d profils de substitution nucléotidique prédits à partir des génomes de référence de deux souches différentes DE B. vulgatus ATCC 848239 et mpk40., En c et en d, des substitutions nucléotidiques ont été identifiées par rapport à un seul gène de référence 16S pour B. vulgatus ATCC 8482 (gène NCBI ID 5304800). Les panneaux gris représentent des régions variables définies par les sites de liaison d’amorce couramment utilisés (tableau supplémentaire 1). Les lignes pointillées indiquent la proportion attendue de substitutions nucléotidiques, étant donné qu’il existe sept copies du gène 16S dans chaque génome., Les données sources sont fournies sous forme de fichier de données Source
profitant du fait que Bacteroides vulgatus était présent en abondance relative élevée dans deux de nos échantillons de microbiome intestinal humain, nous avons ensuite demandé si la variation intragénomique entre les copies du gène 16S pouvait être détectée in vivo. Nous avons aligné chaque séquence pleine longueur classée comme appartenant à notre B. vulgatus V1–V9 OTUs (Fig. 3b et données supplémentaires 1) à une seule séquence génique représentative de B. vulgatus 16S. Nous avons ensuite comparé les profils de substitution nucléotidique résultants (Fig., 3c) avec des profils prédits à partir de deux génomes de référence présents dans la base de données NCBI Refseq19 (fig. 3d).
la majorité de la variation nucléotidique présente dans notre OTU de B. vulgatus in vivo reflète la variation réelle attribuable aux polymorphismes intragénomiques. En revanche, la variation probablement due à des erreurs de séquençage semblait faible et bien inférieure à la fréquence minimale de ~14% qui serait attendue s’il y avait une seule souche de B. vulgatus dans chaque échantillon avec sept copies du gène 16S dans son génome (Fig. 3C, lignes pointillées).
bien que nous ne connaissions pas le vrai nombre de B., les souches de vulgatus présentes dans chaque échantillon in vivo ont montré que les deux profils de substitution de nucléotides ressemblaient davantage à la souche ATCC 8482 qu’à la souche mpk. Il existait également des variations à des locus spécifiques qui pourraient potentiellement indiquer des différences significatives entre les génomes de référence in vivo et ATCC 8482. Par exemple, un seul polymorphisme a été détecté dans la région V5 D’ATCC 8482, qui était présent dans trois copies 16S (43%). Dans le premier échantillon in vivo (Scott), ce polymorphisme était présent dans 84% des lectures, alors que dans le second (IronHorse), il était présent dans 69% des lectures., Ces nombres correspondent étroitement aux nombres attendus si un polymorphisme était présent six et cinq des sept gènes 16S, respectivement.
En conclusion, nous montrons que le séquençage 16S complet du microbiome intestinal humain peut résoudre avec précision les substitutions d’un seul nucléotide qui reflètent la variation intragénomique entre les copies du gène 16s. La présence d’une telle variation indique que les séquences 16S doivent être regroupées pour refléter des unités taxonomiques significatives., En utilisant des OTUs groupés à 99% d’identité, nous montrons que 16S pleine longueur a le potentiel de fournir une résolution taxonomique au niveau des espèces et même des souches. L’analyse des communautés microbiennes à ces niveaux taxonomiques promet de fournir une perspective très différente de celle offerte par les estimations de l’abondance au niveau du genre.
les polymorphismes 16s Intragénomiques sont très répandus
Après avoir démontré qu’il est possible de résoudre les variants de copie intragénomiques in vivo, nous avons ensuite cherché à établir dans quelle mesure de tels variants de copie apparaissent dans les taxons couramment trouvés dans le microbiome intestinal humain., Nous avons également cherché à déterminer si de tels profils peuvent être utilisés régulièrement pour distinguer les souches d’une même espèce.
Nous avons cultivé 381 taxons à partir du microbiome intestinal des individus en bonne santé représentés à la Fig. 3, ainsi que d’autres personnes participant à la même étude originale20 (données supplémentaires 2). Nous avons ensuite effectué un séquençage complet du gène 16S sur des isolats et des lectures séquencées alignées pour identifier les substitutions nucléotidiques caractéristiques des variantes de copie du gène 16S intragénomiques.,
la classification taxonomique des isolats a identifié 58 espèces présumées (données supplémentaires 2), tandis que le regroupement d’une seule séquence représentative pour chaque isolat à une similitude de 99% a donné lieu à 61 Otu (avec entre 1 et 73 isolats attribués à chaque OTU). Au total, 349 des 381 isolats séquencés (54 des 61 Otu) présentaient un ou plusieurs SNP, indiquant la présence de polymorphismes du gène 16S, et 205 profils SNP uniques ont été identifiés lors de la prise en compte d’une erreur potentielle de séquençage (Fig. 4a et données complémentaires 2).

polymorphismes du gène Intragénomique 16s dans les isolats du microbiome intestinal humain. emplacement des SNP présents dans les gènes 16S d’isolats bactériens cultivés individuellement. Les emplacements SNP ont été identifiés grâce à la mise en phase des séquences de gènes 16S sur toute la longueur générées pour chaque isolat individuel. L’axe des X indique la position le long du gène 16S. L’axe des Y désigne les isolats individuels regroupés en fonction de leur phylogénie inférée. Zone bleu foncé indique l’emplacement d’un polymorphisme., Pour plus de clarté, un maximum de cinq isolats appartenant à la même espèce sont indiqués. Pour plus de détails sur les profils de substitution nucléotidique de tous les isolats séquencés, voir Données supplémentaires 2. B-d exemples de profils de substitution nucléotidique montrant des différences de niveau de souche entre les isolats identifiés comme appartenant à trois espèces bactériennes: B Shigella flexneri; C Bifidobacterium longum; d Collinsella aerofaciens. Pour chaque espèce, deux profils de substitution de nucléotides d’isolats sont présentés; cependant, des exemples supplémentaires peuvent être trouvés dans les données supplémentaires 2., Les isolats ont été identifiés comme appartenant à la même espèce si leurs séquences représentatives ont été assignées à la même OTU lors du regroupement à 99% d’identité de séquence. L’identification taxonomique a été réalisée à L’aide de BLAST pour aligner des séquences représentatives à la base de données Blast NCBI 16S (voir méthodes). Les panneaux gris représentent des régions variables définies par les sites de liaison d’amorce couramment utilisés (tableau supplémentaire 1). Les lignes pointillées indiquent la proportion attendue de substitutions nucléotidiques, compte tenu du nombre de copies de gènes 16S prédites pour chaque génome., Les données sources sont fournies sous forme de fichier de données Source
notamment, la comparaison des profils SNP pour les isolats assignés à la même OTU a fréquemment révélé des différences dans la fréquence des SNP qui suggéraient des différences dans les copies intragénomiques du gène 16S entre des taxons étroitement apparentés. Des exemples de différents profils de substitution sont présentés pour trois taxons (fig. 4b-d), qui suggèrent une variation du niveau de souche comparable à celle que nous avons démontrée en principe pour E. coli (fig. 2b).,
En conclusion, nous montrons que de nombreux membres cultivables du microbiome intestinal humain possèdent fréquemment des polymorphismes du gène 16S qui, lorsqu’ils sont correctement pris en compte, ont le potentiel de résoudre des souches de la même espèce.