A teljes 16S gén biztosítja a jobb rendszertani felbontás
A ~1500 bp 16S rrns gén áll kilenc változó régiók tarkított egész erősen konzervált 16S sorozat (Fig. 1a). A teljes gén szekvenálása eredetileg Sanger szekvenálással történt., Ehhez klónozó génekre volt szükség, klónonként két-három leolvasást generáltak, és korlátozott mintavételi mélységet állítottak elő magas költségek és erőfeszítés mellett. Jelenleg azonban a tanulmányok túlnyomó többsége csak a gén egy részét szekvenciálja, mivel a széles körben használt Illumina szekvenáló platform (nagyobb áteresztőképesség, alacsonyabb költség, csökkentett erőfeszítés a Sangerhez képest) rövid szekvenciákat (≤300 bázis) termel., Különböző kistérség a gént, ezért célzott, kezdve egyetlen változó régiók, mint például a V4 vagy V6-os, három változó régiók, mint például a V1–V3 vagy V3–V5 (használt, az Emberi Microbiome Projekt együtt a 454 szekvenálás platform9).
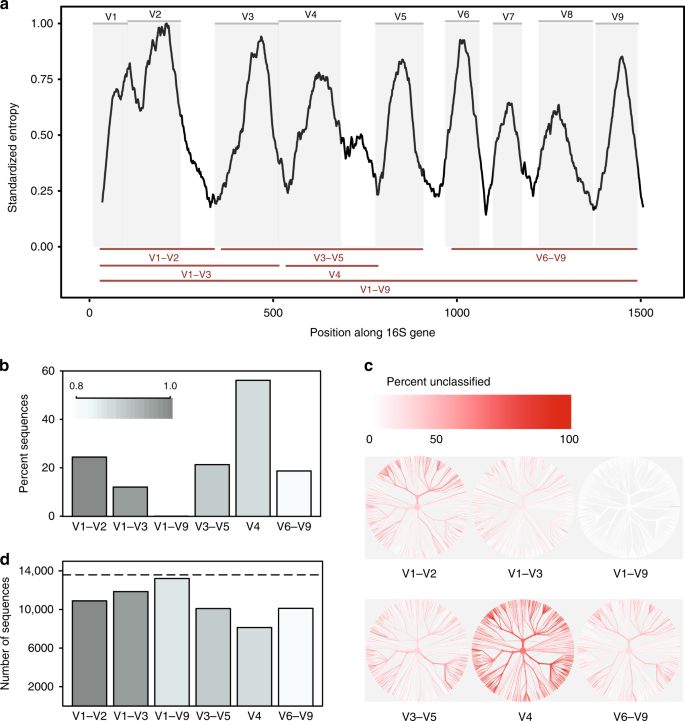
In-silico összehasonlítása 16S rRNA változó régiók. a Shannon entrópia az egész 16S gén összehangolása alapján egyetlen reprezentatív szekvencia minden ismert faj jelen van a Greengenes adatbázisban., A szekvenciákat az Escherichia coli K-12 mg1655 (NCBI gén ID 947777) egyetlen referencia-16S génjéhez igazították. A szürke panelek az általánosan használt primer kötőhelyek által meghatározott változó régiókat ábrázolják (1.Kiegészítő táblázat). A tanulmányban figyelembe vett változó régiók piros vonalakként jelennek meg (alsó). b Aránya szekvenciák minden változó régió, amely nem azonosítható faj szintjén besorolásakor minden sorozat ellen a referencia-adatbázis, amelyből származik, a bizalmi küszöb 80% (RDP osztályozó)., c fák az In-silico adatbázisban található szekvenciák taxonómiája alapján. Ugyanaz a fa biztosított minden változó régióban. Az egyes ágak színe az egyes kládokon belüli szekvenciák arányát tükrözi, amelyeket nem lehetett fajszintre azonosítani. d Az egyes változórégiókhoz tartozó szekvenciák csoportosításakor létrehozott Otu-k száma 99% – os szekvencia-hasonlóság mellett. Szaggatott vonal jelzi az egyedi szekvenciák számát (>1% Különböző) az eredeti adatbázisban., A forrásadatok forrásadatfájlként vannak megadva
azt állítjuk, hogy az alrégiók célzása történelmi kompromisszumot jelent a technológiai korlátozások miatt10. Napjainkban mind a PacBio, mind az Oxford Nanopore szekvenáló platformok képesek rutinszerűen előállítani az 1500 bp-t meghaladó leolvasásokat, és a teljes 16S gén nagy áteresztőképességű szekvenálása egyre inkább elterjedt., Ezért azt javasoljuk, hogy a kompromisszum indoklását újra meg kell vizsgálni, és egy egyszerű In-silico kísérletet végeztünk annak bizonyítására,hogy a teljes hosszúságú 16S szekvenálás előnye az alrégiók célzásával szemben.
letöltöttünk egy sor nem redundáns (azaz > 1% különböző), Teljes hosszúságú 16S szekvenciákat egy nyilvános adatbázisból (Greengenes)., Kihasználva azt a tényt, hogy ezeknek a szekvenciáknak jelentős része beépítette a PCR primer kötőhelyeket, levágtuk őket, hogy Szilikonos amplikonokat generáljanak különböző alrégiókban, a mikrobiómákban általánosan használt PCR primerek elhelyezkedése alapján (ábra. 1a és kiegészítő táblázatok 1-2)., Feltételezve, hogy mindegyik szekvencia a letöltött adatbázis képviselt egyedülálló faj, aztán egy közös osztályozási megközelítés (a Riboszóma Adatbázis Projekt (RDP) classifier11) kiszámításához a gyakoriság, amellyel in-silico amplicons az egyes kistérségi nyújthat pontos, faj szintű rendszertani besorolás (az eredeti adatbázis felhasználásával, mint egy referencia). Egy második kísérletben, az In-silico ampliconjainkat is csoportosítottuk, hogy Otust generáljunk különböző, általánosan használt, szekvencia hasonlósági küszöbértékeken (97%, 98%, 99%).,
megállapítottuk, hogy az alrégiók jelentősen különböztek abban a mértékben, ahogyan magabiztosan megkülönböztethetik a fajok reprezentálására használt Teljes hosszúságú 16S szekvenciákat (ábra. 1b). A V4 régió a legrosszabbul teljesített, az In-silico ampliconok 56% – a nem tudta magabiztosan megfelelni a származás sorrendjének ezen a taxonómiai szinten. Ezzel szemben, amikor egy teljes hosszúságú szekvenciát használtunk az összes változó régióval, szinte minden szekvenciát helyes fajként lehetett osztályozni(kiegészítő ábra. 1a)., Az adatbázisok megváltoztatása és az osztályozási bizalmi küszöbértékek befolyásolták az In-silico ampliconok arányát, amelyek pontosan illeszthetők voltak, de nem befolyásolták az uralkodó trendeket(kiegészítő ábra. 1a, b).
másodszor, a különböző alrégiók elfogultságot mutattak a bakteriális taxonban, amelyet azonosítani tudtak (ábra. 1c). Például a V1-V2 régió rosszul teljesített a phylum Proteobaktériumokhoz tartozó szekvenciák osztályozásakor, míg a V3-V5 régió rosszul teljesített a phylum Actinobaktériumokhoz tartozó szekvenciák osztályozásakor (kiegészítő ábra. 2)., Hasonló tendenciákat figyeltek meg a nemzetség szintjén a lehetséges orvosi relevanciájú taxonok esetében. Bár a teljes V1–V9 régió következetesen gyártott a legjobb eredményt, a V6–V9 régió volt, nevezetesen a legjobb al-régió osztályozása sorozatok tartozó nemzetségek Clostridium, illetve Staphylococcus, a V3–V5 régióban előállított jó eredményeket Klebsiella, a V1–V3 régióban előállított jó eredményeket Escherichia/addigra meglesz (Kiegészítő Ábra. 2 és forrásadatok).
végül az alrégió kiválasztása drámaian befolyásolta az OTUs-ok számát, amikor a-silico-amplikonok csoportosításakor OTUs-t hoztak létre., Amikor fürtözés a 99% – os szekvencia azonosság, mind a kistérség nem sikerült újra a számos különböző szekvenciák jelen az eredeti adatbázis; azonban a V4 régió újra elvégzett legrosszabb (Fig. 1d). Nevezetesen, a relatív száma Ótusz által termelt egyes kistérségi nem volt következetes a különböző identitás küszöbértékek (97%, 98%, 99%, Kiegészítő Ábra. 3), jelezve, hogy a klaszterező algoritmusok viselkedését nehéz lehet megjósolni, ha a szekvenált régióban található információ mennyisége nagyon változó.,
összefoglalva, az alrégiók célzása olyan történelmi kompromisszumot jelent, amely elegendő volt a taxonok nemzetségi szinten vagy annál magasabb szintű azonosításához. Egyszerű In-silico kísérletünk azonban azt mutatja, hogy nem érvényes azt feltételezni, hogy ezen alrégiók egyre finomabb klaszterezése a fajok tükrözéséhez szükséges rendszertani felbontás javulását eredményezi. Bár egyes alrégiók (például a V1–V3) ésszerű közelítést biztosítanak a 16S sokféleséghez, a legtöbb nem rendelkezik elegendő sorrendváltozással a szorosan kapcsolódó taxonok közötti megkülönböztetéshez., Azt is megjegyezzük, hogy a megkülönböztető polimorfizmusokat bizonyos változó régiókra lehet korlátozni; így egyes alrégiók jobban megfelelnek bizonyos taxonok szorosan kapcsolódó tagjainak megkülönböztetésére.
16S génmásolatok tükrözik törzs szintű variáció
csoportosítása 16S szekvenciák OTUs történelmileg szolgált két célra. Először is, eltávolította a kisebb mesterséges szekvencia variánsokat a PCR erősítés és szekvenálási hibák miatt, amikor a szekvenciákat csoportokba csoportosította. Másodszor, összeomlott legitim szekvencia változatok között létezik szorosan kapcsolódó bakteriális taxon., Bár ez utóbbi nem mindig kívánatos, magától értetődik, hogy nem lehet megkülönböztetni a bakteriális taxonokat, amelyek 16S szekvenciái olyan mértékben változnak, amely alacsonyabb, mint egy adott szekvenálási platformon tapasztalt hiba.
a közelmúltban a CCS előrehaladása drámaian javította a hosszú olvasású szekvenálási platformok hibaarányát. Ugyanakkor a számítási módszerek lehetővé tették a legitim vs. mesterséges szekvencia variáció megkülönböztetését., Ezek a technológiai és módszertani fejlődés azt jelenti, hogy a kutatók most már képesek nagy áteresztőképességű szekvenálást végezni, amely pontosan képes kimutatni az egy nukleotid variánsokat az egész 16S génben.
bár csábító feltételezni, hogy az egy nukleotid variánsok különálló, szorosan kapcsolódó taxonokat képviselhetnek,óvatosak vagyunk ezzel a túlságosan egyszerű értelmezéssel szemben,mivel sok bakteriális genom tartalmaz több polimorf példányt a 16S gene12, 13, 14., A PacBio CCS szekvenálást egy 36 fajú baktériumfigura-közösségnél végeztük (3. Kiegészítő táblázat és kiegészítő ábra). 4) annak bizonyítására, hogy I.sok baktérium 16S-szekvenciája ugyanazon genomon belüli operonok között változik, és ii. hogy a nagy áteresztőképességű szekvenálás kellően pontos ahhoz, hogy megoldja ezeket az intragenomikus különbségeket.
a Pacsbio teljes hosszúságú 16S szekvenciákat egy referencia adatbázishoz igazítottuk, amely egyetlen reprezentatív 16S szekvenciát tartalmaz a mock közösség minden tagja számára, és az igazítási statisztikákat használtuk ennek a szekvenálási megközelítésnek a pontosságának értékelésére., Összehasonlítva a menetek száma létrehozásához használt a CCS az esemény egyetlen nukleotid csere, betoldások, valamint a törlések jelezte, hogy tíz halad is csökkenti ezeket a kombinált hibát, hogy egy minimális frekvencia < 1.0% (bár figyelemre méltó, hogy a minimális elérhető hiba változatos közötti sorrend fut; Kiegészítő Ábra. 5). Ugyanakkor megfigyeltük a törlési hibák egybeesését a homopolimer futásának helyével a referenciaszekvenciáinkban(kiegészítő ábra., 6), amely nem volt nukleotidspecifikus, és súlyosbította a szekvenált homopolimer hossza (kiegészítő ábra. 7). Ezt követően az Escherichia coli 16S génben az Illumina teljes genom (WGS) szekvenálásával validáltuk a törléseket, amelyek kimutatták, hogy a PacBio szekvenciákban előforduló törlések közül csak az egyik valódi volt (kiegészítő ábra. 8).,
meggyőződve arról, hogy a CCS szekvenálás képes 16S olvasás alacsony frekvenciájú helyettesítési hibák, mi a következő indokoltan, hogy egy részét a helyettesítési hibák belül pontosan igazított olvas tükröznie kell variáció tulajdonítható 16S polimorfizmusok egy faj genome12. Például az E. coli K-12 alcsoporthoz igazodik. Az MG1655 szubsztitúciós profilt mutatott, amely pontosan azt tükrözte, amelyet az ebben a genome15-ben ismert 16S szekvenciák mind a hétének összehangolásával jósoltak (ábra. 2a, c)., Ezen nukleotid szubsztitúciók sztöchiometriáját tovább tudtuk érvényesíteni az összehasonlíthatóan igazított Illumina WGS olvasás variációjának számszerűsítésével (ábra. 2b) és bizonyítsa, hogy egy hasonló helyettesítési profil több szekvenálási folyamaton is reprodukálható volt (kiegészítő ábra. 9)., Igazítás más referencia szekvenciák a mi mock közösség mutatott hasonló tendencia bőséges szubsztitúciók lokalizált specifikus bázis pozíciókat mentén 16S gén, bár megjegyezzük, hogy a jel-zaj arány jelentősen nőtt, ha a 16S gén kérdéses volt kevesebb, mint 100 igazított olvasás (kiegészítő ábra. 10).

polimorfizmusok az E. coli 16S rRNA génszekvenciákban. a helyettesítések helyzete és gyakorisága az E-ben., coli törzs K-12 MG1655 v1–V9 ampliconok, amelyeket a pacbio RS II platformon szekvenáltak. b az Illumina MiSeq platformon az izolált E. coli K-12 mg1655 törzs genomikus szekvenálásából eredő szubsztitúciók helyzete és gyakorisága. A nagyított régiók megfelelő pozíciókat mutatnak az E. coli K-12 MG1655 referencia genomban jelen lévő mind a hét 16S gén összehangolásában. Az RRND operon (**) 16S szekvenciáját használják referenciaként az összes SNP-fázishoz. c az e várható nukleotid szubsztitúciós profilja., coli K-12 MG1655 alapján összehangolása a hét 16S génszekvenciák jelen a referencia Genom. d az E. coli O157 Sakai várható helyettesítési profilja a referencia genomban jelen lévő hét 16S génszekvencia összehangolása alapján. A szürke panelek az általánosan használt primer kötőhelyek által meghatározott változó régiókat ábrázolják (1.Kiegészítő táblázat). A szaggatott vonalak jelzik a nukleotid szubsztitúciók várható arányát, mivel minden genomban hét 16S génmásolat található., A forrásadatok Forrásadatfájlként vannak megadva
az a megfigyelés, hogy a hosszú olvasású szekvenálás képes azonosítani a 16S polimorfizmusokat ugyanazon genomban, fontos következményekkel jár. Először is, azt mutatja,hogy nem érvényes azt feltételezni, hogy a nagy áteresztőképességű szekvencia egy vagy néhány nukleotidtól eltérő, különálló taxa6, 16. Egyetlen genomban két vagy több 16S szekvencia azonos lehet, míg mások egyediek lehetnek., Ennek megfelelően néhány homológ 16S loci megtarthatja az azonos szekvenciát két szorosan kapcsolódó törzs között, míg mások eltérhetnek egy vagy néhány nukleotidpozícióban. Ebben az összefüggésben minden olyan közösségi szintű vagy rendszertani értelmezése 16S adatok ideális esetben figyelembe azt a tényt, hogy a relatív rengeteg 16S sorozatok eredő nagyon közeli rokon taxonok tükrözi egy lineáris kombinációja (i) a frekvencia, amellyel az egyes egyedi szekvencia képviselt egész genom szekvenciát jelent, valamint (ii) a relatív abundancia a genom szekvenciát jelent minden taxon.,
második, bár az intragenomikus 16S szekvencia-variáció bonyolítja a közösségi szintű elemzést, a 16S gén teljesítményének növelése is lehetővé teszi a szorosan kapcsolódó taxonok közötti megkülönböztetést, mivel lehetővé teszi a szekvencia-alapú összehasonlítást több eltérő loci-n keresztül. Például elegendő nukleotid variáció létezik ahhoz, hogy megkülönböztessük az E. coli K-12 mg1655 törzset az O157 Sakai enterohemorrhagiás törzstől (ábra). 2c, d)., Így azt állítjuk, hogy megfelelő elszámolás esetén a többszörös polimorf 16S másolatok nem jelentenek kellemetlenséget, hanem lehetővé teszik a 16S gén használatát a törzsszintű mikrobióma-elemzésben. Azt is megjegyezzük, hogy az intragenomikus 16S szekvencia variáció ereje a szorosan kapcsolódó taxonok megkülönböztetésére valószínűleg csökken, ha részleges 16S szekvenciákat használnak. Például az SNP-k megkülönböztetik a K-12 mg1655 E. coli törzseket (ábra. 2C) O157 Sakai (ábra. 2d) a V1, V2, V6 és V9 változó régiókban találhatók.,
16S a polimorfizmusok in vivo megoldhatók
a Mikrobiómaközösségek gyakran összetettek, különböző biokémiai környezetben léteznek (pl. széklet, nyál, köpet stb.) és több száz egyedi taxont tartalmaz, amelyek relatív bősége széles dinamikatartományt ölel fel. Ez a komplexitás nem képviselteti magát sem in-silico, sem mock közösségi kísérletekben. Ezért további kísérletet végeztünk annak bizonyítására, hogy a teljes 16S gén szekvenálása, miközben az intragenomikus 16S SNP-k elszámolása képes megoldani a szorosan kapcsolódó bakteriális taxonokat in vivo.,
elvégeztük a V1–V9 régió PacBio CCS szekvenálását négy egészséges felnőtt önkéntesektől gyűjtött emberi székletmintára. Összehasonlításképpen a V1–V3 régiót az Illumina MiSeq segítségével szekvenáltuk, és a fajszintű taxonómiai számszerűsítés viszonyítási alapjaként metagenomic WGS (mWGS) szekvenálást végeztünk az Illumina NextSeq segítségével. Annak felmérése érdekében, hogy ezek a szekvenálási megközelítések milyen mértékben képesek megoldani a szorosan kapcsolódó taxonokat, a Bacteroides nemzetségre összpontosítottunk., Amellett, hogy bőséges az emberi bélben, ez a nemzetség rendkívül változatos, több fajt tartalmaz, amelyek mind jó, mind rossz hatással lehetnek az emberi egészségre17. Korábban modell taxonként is használták a 16S gén hasznosságának bizonyítására a nagy felbontású taxonómiai elemzésekhez18.
amikor a Bacteroides bőséget a nemzetség szintjén számoltuk, a v1–V9 szekvenálás és a V1–V3 szekvenálás hasonló eredményeket hozott., Mindkét megközelítés két alacsony Bacteroides relatív bőség (~10-25%) és két magas Bacteroides relatív bőség (~40-60%; ábra. 3a). Az mwgs-szekvenálással végzett fajszintű számszerűsítés azonban sokkal nagyobb sokszínűséget tárt fel, az egyes egyedek bélében eltérő Bacteroides fajok dominálnak (ábra. 3b és kiegészítő adatok 1). Amikor a vidrákat 99% – os identitás mellett csoportosították, mind a V1–V9, mind a V1-V3 szekvenálás képes volt tükrözni ezt a fajszintű variációt(ábra., 3b), azzal a figyelemre méltó kivétellel, hogy a V1-V3 szekvenálás nem észlelte a Bacteroides intestinalis-t, amely bőséges volt a négy emberi bél mikrobióma mintájának egyikében. Ezen eredmények alapján arra a következtetésre jutunk, hogy ha megfelelő identitási küszöbértékkel (például 99%-kal) együtt alkalmazzák, az OTU-alapú megközelítések képesek megoldani az emberi bélben megfigyelt fajszintű sokféleséget. Megjegyezzük továbbá, hogy bár a teljes hosszúságú 16S szekvenálás optimális lehet A fajszintű elemzéshez, a rendkívül informatív változó régiók (például V1–V3) szintén megfelelőek lehetnek erre a célra.,

Bacteroides kimutatása emberi székletmintákban. a Bacteroides nemzetség relatív bősége négy emberi székletmintában, v1–V9 ampliconok (x-tengely) vagy V1–V3 ampliconok (y-tengely) alkalmazásával számszerűsítve. b A Bacteroides fajok relatív bősége ugyanabban a négy mintában. A fajok bőségét mwgs szekvenálásból vagy V1–V3/V1–V9 OTUs-ból számszerűsítették, 99% – os identitással., A bőség az mWGS által számszerűsített legnagyobb mennyiségben előforduló fajok esetében jelenik meg (az egyes platformok által észlelt összes Bacteroides faj bőség-becslését lásd az 5.Kiegészítő táblázatban). C nukleotid szubsztitúciós profilok által generált összehangolása minden v1-V9 amplicon szekvenciák rendelt egyetlen OTU azonosított Bacteroides vulgatus. A két, nagy B. vulgatus relatív bőség (IronHorse és Scott) arányú székletminta profiljai láthatók. két különböző B. vulgatus törzs, az ATCC 848239 és az mpk40 referencia genomjaiból jósolt nukleotid szubsztitúciós profil., Mind a c, mind a d esetében nukleotid szubsztitúciókat azonosítottak a B. vulgatus ATCC 8482 (NCBI gén ID 5304800) egyetlen referencia-16S génjéhez képest. A szürke panelek az általánosan használt primer kötőhelyek által meghatározott változó régiókat ábrázolják (1.Kiegészítő táblázat). A szaggatott vonalak jelzik a nukleotid szubsztitúciók várható arányát, mivel minden genomban hét 16S génmásolat található., A forrásadatokat forrásadatként adjuk meg
kihasználva azt a tényt, hogy a Bacteroides vulgatus nagy relatív bőségben volt jelen két emberi bél mikrobiómamintánkban, majd megkérdeztük, hogy a 16S génmásolatok közötti intragenomikus variáció kimutatható-e in vivo. A B. vulgatus V1-V9 OTUs–hoz tartozó összes Teljes hosszúságú sorozatot igazítottuk (ábra. 3b és kiegészítő adatok 1) egyetlen reprezentatív B. vulgatus 16S génszekvenciához. Ezután összehasonlítottuk a kapott nukleotid szubsztitúciós profilokat (ábra., 3c) az NCBI RefSeq adatban19-ben található két referencia genomból jósolt profilokkal (ábra. 3d).
az in vivo generált B. vulgatus OTU-ban jelen lévő nukleotid variációk többsége az intragenomikus polimorfizmusoknak tulajdonítható valódi variációt tükrözte. Ezzel szemben a szekvenálási hibák miatt valószínű variáció alacsonynak és jóval a minimális ~14% – os gyakoriság alatt jelent meg, amely akkor várható, ha minden mintában egyetlen B. vulgatus törzs lenne, hét 16S gén másolattal a genomjában (ábra. 3C, szaggatott vonalak).
bár nem tudtuk a B valódi számát., az egyes in vivo mintákban jelen lévő vulgatus törzsek figyelemre méltóak voltak, hogy mindkét nukleotid szubsztitúciós profil közelebb állt az ATCC 8482 törzshez, mint az mpk. Az in vivo és az ATCC 8482 referencia-genomok közötti jelentős eltéréseket potenciálisan jelző lokális variációk is léteztek. Például egyetlen polimorfizmust észleltek az ATCC 8482 V5 régiójában, amely három 16S példányban volt jelen (43%). Az első in vivo mintában (Scott) ez a polimorfizmus az olvasás 84% – ában volt jelen, míg a másodikban (IronHorse) az olvasás 69% – ában volt jelen., Ezek a számok szorosan megfelelnek a várt számoknak, ha a polimorfizmus jelen volt a hét 16S gén közül hat, illetve öt.
összefoglalva, megmutatjuk, hogy az emberi bél mikrobióma teljes hosszúságú 16S szekvenálása pontosan megoldhatja az egy nukleotid szubsztitúciókat, amelyek tükrözik az intragenomikus variációt a 16S génmásolatok között. Az ilyen variációk jelenléte azt jelzi, hogy a 16S szekvenciákat csoportosítani kell, hogy tükrözzék az értelmes taxonómiai egységeket., A 99%-os identitás mellett csoportosított OTUs használatával megmutatjuk, hogy a teljes hosszúságú 16-ok képesek fajokat, sőt törzs szintű taxonómiai felbontást biztosítani. A mikrobiális közösségek elemzése ezen rendszertani szinteken azt ígéri,hogy nagyon eltérő perspektívát biztosít a nemzetség szintű bőség becslései által.
Intragenomic 16 polimorfizmusok rendkívül elterjedt
, Amelyek bizonyították, hogy lehetséges, hogy megoldja intragenomic másolás változatok in vivo, mi a következő igyekezett megállapítani, hogy milyen mértékben az ilyen másolatot változatok jelennek meg, a fajok gyakran megtalálható az emberi bélben microbiome., Arra is törekedtünk, hogy megállapítsuk, hogy az ilyen profilok rutinszerűen használhatók-e ugyanazon faj törzseinek megkülönböztetésére.
381 taxát tenyésztettünk az ábrán látható egészséges egyének bél mikrobiómájából. 3, valamint más, ugyanazon eredeti tanulmányban20 részt vevő személyektől (2.Kiegészítő adat). Mi ezt követően végeztek teljes hossza 16 gén szekvencia a külön rendezi szekvenált olvassa azonosítani nukleotid csere jellemző intragenomic 16 gén másolás változatok.,
Rendszertani besorolása izolátumok azonosított 58 vélt fajok (Kiegészítő Adatok 2), míg a klaszter egyetlen képviselője sorozat minden izolátum a 99% – os hasonlóságot eredményezett 61 Ótusz (a között 1 73 izolátumok hozzárendelt OTU). Összesen 349 381 szekvenált izolátumok (54 61 Ótusz) volt egy vagy több SNP, jelezve, hogy a jelenléte 16 gén polimorfizmusok, valamint 205 egyedi SNP profilok azonosítottak, amikor számviteli potenciális sorrendi hiba (Fig. 4a és kiegészítő adatok 2).

Intragenomic 16S gene polymorphisms in human gut microbiome isolates. az SNP-k helye az egyedileg tenyésztett bakteriális izolátumok 16S génjeiben található. Az SNP helyeket az egyes izolátumok számára létrehozott teljes hosszúságú 16S génszekvenciák fokozatos alkalmazásával azonosították. Az X-tengely a 16S gén mentén helyezkedik el. Az Y-tengely az egyes izolátumokat csoportosítja a termékezett filogenitásuk alapján. A sötétkék régió jelzi a polimorfizmus helyét., Az egyértelműség érdekében legfeljebb öt, ugyanahhoz a fajhoz tartozó izolátum látható. Az összes szekvenált izolátum nukleotid szubsztitúciós profiljának részleteiről lásd a 2. Kiegészítő adatot. B-d példák a nukleotid szubsztitúciós profilokra, amelyek törzsszintű különbségeket mutatnak a három baktériumfajhoz tartozó izolátumok között: B Shigella flexneri; c Bifidobacterium longum; D Collinsella aerofaciens. Minden faj esetében két izolált nukleotid szubsztitúciós profil látható; további példák azonban a 2. Kiegészítő adatokban találhatók., Az izolátumokat ugyanazon fajhoz tartozóként azonosították, ha reprezentatív szekvenciáikat ugyanahhoz az OTU-hoz rendelték, amikor 99% – os szekvenciaazonosság mellett csoportosultak. A taxonómiai azonosítást BLAST segítségével végezték el, hogy a reprezentatív szekvenciákat az NCBI 16S BLAST adatbázishoz igazítsák (lásd a módszereket). A szürke panelek az általánosan használt primer kötőhelyek által meghatározott változó régiókat ábrázolják (1.Kiegészítő táblázat). A szaggatott vonalak jelzik a nukleotid szubsztitúciók várható arányát, figyelembe véve az egyes genomokra előrejelzett 16S génmásolatok számát., A forrásadatokat forrásadatfájlként adják meg
nevezetesen, az ugyanazon OTU-hoz rendelt izolátumok SNP-profiljainak összehasonlítása gyakran kimutatta az SNP-k gyakoriságának különbségeit, amelyek az intragenomikus 16S génmásolatok közötti különbségekre utaltak a szorosan kapcsolódó taxonok között. Példák a különböző helyettesítési profilok jelennek meg három taxon (ábra. 4B-d), amelyek olyan törzsszintű változásra utalnak, amely összehasonlítható az E. coli esetében elvben bemutatott változással (ábra. 2b).,
összefoglalva, megmutatjuk, hogy az emberi bél mikrobióma sok kulturálható tagja gyakran rendelkezik 16S gén polimorfizmusokkal, amelyek megfelelő elszámolás esetén képesek ugyanazon faj törzseinek megoldására.