RNS szekvenálás (Wang 2009) bevezetése számos laboratóriumban gyorsan felváltja a génexpressziós mikroarraysokat. Az RNS-seq segítségével számszerűsítheti, felfedezheti és profilozhatja az RNS-eket. Ehhez a technikához az mRNS-t (és más RNS-t) először cDNA-ra alakítják át. Ezután a cDNA-t használják bemenetként egy következő generációs szekvenáló könyvtár előkészítéséhez. Ebben a cikkben röviden áttekintem az RNS-seq-t, és bemutatom a ma alkalmazott főbb módszereket.
miért “jobb” az RNS-Seq, mint a Mikroarrays?,
számos előnye van az RNS-seq-nak a mikroarrays-szal szemben:
az RNS-seq-vel több, mint a differenciál gén expressziója. Bár vannak mikroarrays elérhető exon-level és microRNA elemzés, a legtöbb felhasználó még mindig érdekli az alapvető, valószínűleg 3 ” elfogult, differenciál gén expresszió. Az RNS-seq segítségével megnézheti a kódoló és nem kódoló RNS-t, a splicing és allélspecifikus expressziót, és valószínűleg hamarosan a teljes hosszúságú cDNS-szekvenciákat, kiküszöbölve az izoformák következtetésének vagy összeállításának szükségességét.,
A Mikroarrays is elfogult, mivel el kell döntenünk, hogy milyen tartalmat kell elhelyezni a tömbben. Mivel az RNS-seq nem használ szondákat vagy primereket, az adatok sokkal alacsonyabb torzításokat szenvednek (bár nem azt akarom mondani, hogy az RNS-seq-nek nincs).
az RNS-seq digitális adatokat biztosít igazított olvasási számok formájában, ami nagyon széles dinamikus tartományt eredményez, javítva a ritka átiratok észlelésének érzékenységét.
Ez is nagyon költség-versenyképes mikroarrays, mint ma, között 6-30 mintát lehet multiplexelni egyetlen Illumina szekvenáló sávban.,
végül újraelemezheti az RNS-seq adatkészletet, mivel több információ áll rendelkezésre a transzkriptómáról. Ha egy olyan tanulmány jelenik meg, amely egy érdekes splice-változatot mutat be egy hasonló rendszerben, mint amelyen dolgozik, akkor érdemes visszamenni, és megnézni a mintákban lévő splicinget; és már rendelkezne az adatokkal.
hogyan fejti ki hatását az RNS-Seq?
számos módszer létezik RNS-seq kísérlet elvégzésére. Valójában a technikák olyan gyorsan fejlődnek, hogy nehéz lehet eldönteni, hogy melyiket használja., Az alapvető választás között 1) random-primed cDNA szintézis kettős szálú cDNA vagy 2) RNS-ligációs módszerek (felül, összehasonlítva Levin 2010). A legtöbb ember használja az első módszert, majd további választást kell tennie egy szálspecifikus protokoll és egy olyan között, amely nem. A laboratóriumomban leginkább alkalmazott módszer az Illumina TruSeq RNS-seq, amely egy random-primed cDNA szintézis nem-szál-specifikus protokoll.
miután van egy szekvenáló könyvtár, akkor szekvenált egy meghatározott mélységbe, amely attól függ, hogy mit szeretne csinálni az adatokat., Ezek a leolvasások igazodnak a genomhoz vagy a transzkriptómához, és számítanak a differenciál gén expressziójának meghatározására vagy további elemzésre a splicing és az izoform expresszió meghatározására. A legtöbb ember az RNS szekvenálását páros végű 50-100bp módszerekkel végzi. A kivétel a microRNA szekvenálás, mivel ez a legtöbb esetben csak egyvégű 36bp szekvenálást igényel.
RNS-Seq módszerünk
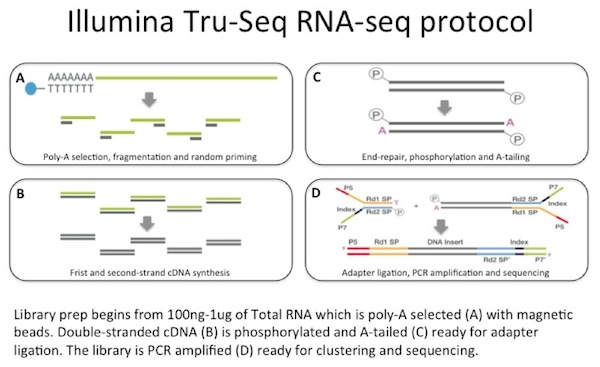
100 ng-1 µg teljes RNS-t használunk az oligo-dT bevont mágneses gyöngyökkel történő mRNS-rögzítés bemeneteként. Az mRNS fragmentált, majd egy véletlen alapozású cDNA szintézist hajtanak végre., A kapott kétszálú cDNA használják a bemenet egy szabványos Illumina library prep, amely magában foglalja a vég-javítás, adapter ligálás és PCR erősítés, hogy kapsz egy könyvtár kész szekvenálás.
miért zavarja a Szálinformációkat?
sok vita folyt az érzékellenes transzkripcióról és annak biológiai jelentőségéről. Ha érdekli az egyszerű differenciál génexpresszió, akkor a szálinformációk nem adnak sokat a kísérlethez, de összetettebbé teszik a protokollt., Miután elmondta, hogy a legszélesebb körben elfogadott módszert túl sok extra erőfeszítés nélkül hajthatja végre. Ehhez a 2. szál cDNA szintézis során használja az uracil-t a timin helyett. Kövesse az Illumina könyvtár prep, mint a normál, de miután adapter ligálás előtt PCR amplifikáció add uracil-DNS glikoziláz lebontani a 2ndstrand. Ez azt eredményezi, hogy az összes olvasás ugyanabban az orientációban kezdődik, így meghatározhatja, hogy melyik szálat írják át a mintában.
mit tehetsz valójában az RNS-Seq – val?,
az RNS-seq egy hatékony és sokoldalú eszköz, amelyet az elmúlt években széles körben publikáltak. Kiválasztottam néhány kedvencemet (néhányat az általam kezelt központi létesítményben végzett munkából), hogy bemutassam, mit tehet az RNS-szekvenálással.
- Jabbari, et al. használt RNS-seq, hogy vizsgálja meg a pikkelysömör és új gének funkcionális analízis. Összehasonlították RNS-seq-adataikat a közzétett tömbkutatásokkal, és 1700 új jelöltet találtak. Ezeket a qPCR validálta, és a psoriasis funkcionális adatbázisaival való összehasonlítás támogatta a patogenezisben betöltött szerepüket.
- Kutter, et al., használt RNS-seq egy tanulmány keresi a természetvédelmi az RNS Polimeráz III kötelező emlősök, hogy érvényesítse kifejezés a gének által elfoglalt Pol III., mint a mintájukat által ChIP-seq.
- Mercer, et al. kombinált RNS-seq és mikroarray-alapú rögzítés a ritka átiratok azonosítására és jellemzésére, amelyek általában nem észlelhetők. A célzott átiratok sorrendben növekedtek az olvasási bőség 0, 21%-ról a rögzítés előtti 80% – ra., Több mint 200 korábban nem jelölt izoformát találtak közel 50 fehérje-kódoló loci-ra, köztük egy új alternatív TP53 izoformát, amely nagyon jól jellemzett gén. Ez arra utal, hogy a genomban és a transzkriptómában még mindig sok a bonyolultság, amit meg kell oldani.
összefoglalva, az RNS-seq továbbra is fejlődő eszköz, de a legtöbb esetben előnyösebb a mikroarraysnál. Érzékenyebb, robusztusabb és költséghatékonyabb is lehet. Milyen RNS-seq projekteket tervez most a projekthez?,
jabbari et al: a pikkelysömör transzkripciós profilozása RNS-seq segítségével korábban azonosítatlan, Differenciálisan expresszált géneket tár fel. Journal of Investigative Dermatology 2011.
Kutter et al: A Pol III hat emlősben való kötődése az aminosav-izotípusok közötti megőrzést mutatja, annak ellenére, hogy a tRNS gének eltérnek egymástól. Nature Genetics 2011.
Levin et al: a szálspecifikus RNS szekvenálási módszerek átfogó összehasonlító elemzése. Természet Módszerek 2010.
Mercer et al: a célzott RNS szekvenálás feltárja az emberi transzkriptóma mély összetettségét. Nature Biotechnology 2012.,
Wang et al: RNS-Seq: forradalmi eszköz a transzkriptomika számára. Nat Rev Genet 2009.
eredetileg 2012.május 25-én jelent meg. 2015.augusztus 16-án frissítve és felülvizsgálva.

Ez segített neked? Akkor kérjük, ossza meg a hálózat.