full 16S genet gir bedre taksonomisk oppløsning
~1500 bp 16S rRNA-genet består av ni variable regioner ispedd hele svært bevart 16S sekvens (Fig. 1a). Sekvensering av hele genet ble opprinnelig utført av Sanger-sekvensering., Dette kreves for kloning av gener, generere og montering av to til tre leser per klone, og produserer begrenset prøvetaking dybde på høy kostnad og innsats. For øyeblikket, men det store flertallet av studier sekvens bare en del av genet, fordi mye brukt Illumina-sekvensering plattform (høyere gjennomstrømning, lavere kostnader, redusert innsats sammenlignet med Sanger) produserer korte sekvenser ( ≤ 300 baser)., Ulike sub-regioner av genet er derfor målrettet, alt fra enkle variable regioner, som for eksempel V4 eller V6, tre variable regioner, som for eksempel V1–V3 eller V3–V5 (brukes i Human Microbiome Project i forbindelse med 454-sekvensering platform9).
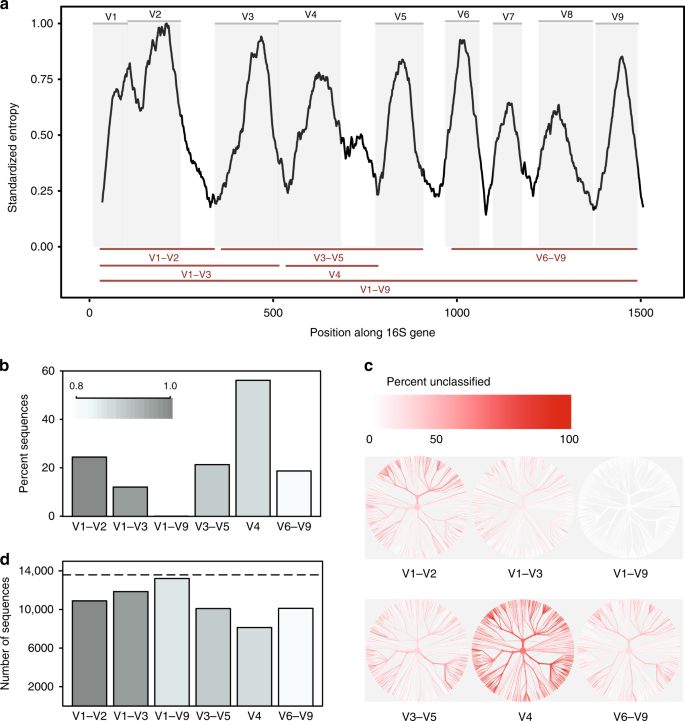
I-silico sammenligning av 16S rRNA-variable regioner. en Shannon entropi over 16S gen basert på plasseringen av en enkelt representant sekvens for hvert kjente arter til stede i Greengenes database., Sekvenser var innrettet mot en enkelt referanse 16S genet for Escherichia coli K-12 MG1655 (NCBI Genet ID 947777). Grå paneler skildre variable regioner definert av brukte primer-bindende områder (Supplerende Tabell 1). Variable regioner vurdert i denne studien er vist som røde linjer (nederst). b Andel av sekvenser for hver variabel regionen som ikke kunne identifiseres til art nivå når klassifisere hver sekvens mot referanse-database der det ble hentet på en tillit terskelen til 80% (RDP classifier)., c Trær basert på forskjellige typer sekvenser til stede i i-silico database. Det samme treet er gitt for hver variabel regionen. Fargen på hver gren, og reflekterer andelen av sekvenser innenfor hver clade som ikke kunne identifiseres til art nivå. d antall OTUs opprettet når clustering sekvenser for hver variabel regionen på 99% sekvens likhet. Stiplet linje angir antall unike sekvenser (>1% annet) i den opprinnelige databasen., Kilde data er gitt som en Kilde Data file
Vi hevde at målretting sub-regioner representerer et historisk kompromiss, på grunn av teknologien restrictions10. I dag, både PacBio og Oxford Nanopore sekvensering plattformer er i stand til rutinemessig å produsere leser i overkant av 1500 bp og høy gjennomstrømning sekvensering av hele 16S genet blir stadig mer utbredt., Vi vil derfor foreslå at begrunnelsen for dette kompromisset trenger å bli tolket og vi utført en enkel in-silico eksperiment for å demonstrere fordel av full-lengde sekvensering av 16S over målretting av sub-regioner.
Vi lastet ned et sett av ikke-redundant (dvs., > 1% annet), full-lengde 16S sekvenser fra en offentlig database (Greengenes)., Tar nytte av det faktum at en betydelig andel av disse sekvensene innlemmet PCR-primer-bindende områder, vi trimmet dem til å generere i-silico amplikoner for ulike sub-regioner, basert på plasseringen av PCR-primere som vanligvis brukes i microbiome studier (Fig. 1a og Supplerende Tabeller 1-2)., Forutsatt at hver sekvens i vår database lastet ned representerte en unik arter, vi brukte en felles klassifisering tilnærming (Ribosomet Database-Prosjektet (RDP) classifier11) til å beregne frekvens som i-silico amplikoner for hver sub-regionen kunne gi nøyaktig, arter-nivå taksonomisk klassifisering (ved hjelp av den opprinnelige databasen som referanse). I et annet eksperiment, vi har også gruppert våre silico amplikoner for å generere OTUs på ulike, ofte brukt, sekvens likhet tersklene (97%, 98%, 99%).,
Vi fant ut at sub-regioner skilte seg vesentlig i den grad de kan trygt diskriminere mellom full-lengde 16S sekvenser som brukes til å representere arter (Fig. 1b). V4-regionen utført verste, med 56% av i-silico amplikoner sviktende trygt å matche deres sekvens av opprinnelse på dette taksonomisk nivå. Når derimot en full-lengde sekvens med alle variable regioner ble brukt, var det mulig å klassifisere nesten alle sekvenser som riktig arter (Supplerende Fig. 1a)., Endre databaser og klassifisering tillit terskler påvirket andelen av i-silico amplikoner som kan være nøyaktig matchet, men det hadde ingen innvirkning rådende trender (Supplerende Fig. 1a, b).
Andre, forskjellige sub-regioner viste skjevhet i den bakterielle taksa de var i stand til å identifisere (Fig. 1c). For eksempel, den V1–V2-regionen utført dårlig på å klassifisere sekvenser som tilhører den phylum Proteobacteria, mens V3–V5-regionen utført dårlig på å klassifisere sekvenser som tilhører den phylum Actinobacteria (Supplerende Fig. 2)., Lignende trender ble sett på slekten nivå for taksa av potensielle medisinsk relevans. Selv om full V1–V9-regionen konsekvent produsert de beste resultatene, V6–V9-regionen var særlig den beste sub-region for klassifisering av sekvenser som tilhører slektene Clostridium og Staphylococcus, den V3–V5-regionen har gitt gode resultater for Klebsiella, og V1–V3-regionen har gitt gode resultater for Escherichia/Shigella (Supplerende Fig. 2 og Kilde Data).
til Slutt, valg av sub-region dramatisk påvirket antall OTUs dannet når clustering i-silico amplikoner å skape OTUs., Når clustering på 99% sekvens identitet, alle sub-regioner klarte ikke å gjenskape antall forskjellige sekvenser til stede i den opprinnelige databasen, men V4-regionen igjen utført verste (Fig. 1d). Spesielt er det relative antallet av OTUs produsert av hver sub-regionen var ikke konsekvent på annen identitet tersklene (97%, 98%, 99%, Supplerende Fig. 3), noe som indikerer at atferden til clustering algoritmer kan være vanskelig å forutsi når mengden informasjon som finnes i en sekvensert regionen er svært variabel.,
I konklusjonen, rettet mot sub-regioner representerer et historisk kompromiss som var tilstrekkelig for identifisering av taksa i slekten nivå eller over. Men, vår enkle i-silico eksperiment viser at det ikke er gyldig grunn til å anta at stadig finere gruppering av disse sub-regioner vil resultere i forbedret taksonomisk oppløsning som er nødvendig for å reflektere arter. Selv om noen sub-regioner (f.eks., V1–V3) gi en rimelig tilnærming av 16S mangfold, de fleste ikke ta tilstrekkelig sekvens variasjon til å diskriminere mellom nært beslektede taksa., Vi ser også at diskriminerende polymorfismer kan være begrenset til bestemte variable regioner, og dermed visse sub-regioner vil være bedre egnet for diskriminering nært knyttet medlemmer av bestemte taksa.
16S genet kopi varianter reflektere sil-nivå variasjon
Gruppering av 16S sekvenser i OTUs har historisk sett tjente to formål. Først, det er fjernet mindre kunstig sekvens varianter på grunn av PCR-amplifikasjon og sekvensering feil når du slår sammen sekvenser i grupper. For det andre har kollapset legitime sekvens varianter som eksisterer mellom nært beslektede bakteriell taksa., Selv om sistnevnte kan ikke alltid være ønskelig, det står til grunn at du ikke kan skille mellom bakteriell taksa som 16S sekvenser varierer på en pris som er lavere enn det feil oppstått på et bestemt sekvensering plattform.
Nylig, fremskritt innen CCS ha dramatisk forbedret feil priser på lenge-les-sekvensering plattformer. På samme tid, beregningsmetoder har gjort det mulig å skille mellom legitime vs. kunstig sekvens variasjon., Disse teknologiske og metodologiske fremskritt mener forskerne nå har potensial til å utføre høy gjennomstrømning sekvensering som kan nøyaktig registrere enkelt-nukleotid varianter over hele 16S-genet.
Selv om det er fristende å anta at enkelt-nukleotid varianter kan representere forskjellige, nært knyttet taksa, vi advare mot denne altfor forenklede tolkning på grunn av det faktum at mange bakterielle genomer inneholde flere polymorfe kopier av 16S gene12,13,14., Vi utført PacBio CCS-sekvensering av en 36 arter bakteriell mock samfunnet (Supplerende Tabell 3 og Supplerende Fig. 4) for å vise (i) at 16S sekvens av mange bakterier varierer mellom operons innenfor samme genom og (ii) at høy gjennomstrømning sekvensering er tilstrekkelig nøyaktig for å løse disse intragenomic forskjeller.
Vi justert PacBio full-lengde 16S-sekvenser til en referanse database som inneholder en enkelt representant 16S sekvens for hvert enkelt medlem av vår mock samfunnet og brukes justeringen statistikk for å vurdere nøyaktigheten av denne sekvensering tilnærming., Å sammenligne antall omganger som brukes til å generere en CCS med forekomst av enkelt-nukleotid erstatninger, innsettinger og slettinger indikert at ti passerer kunne minimere disse kombinert feil til et minimum frekvens av < 1.0% (selv om det var kjent at minimum oppnåelig feil variert mellom sekvensering går; Supplerende Fig. 5). Men gjorde vi observere en tilfeldighet sletting feil med plasseringen homopolymer går i vår referanse sekvenser (Supplerende Fig., 6), som ikke var nukleotid-spesifikke og ble forsterket av lengden på sekvensert homopolymer (Supplerende Fig. 7). Vi senere sletting validerte innenfor Escherichia coli 16S-genet ved hjelp av Illumina hele genom hagle (WGS) – sekvensering, som viste at bare ett av slettinger som oppstår i PacBio sekvenser var ekte (Supplerende Fig. 8).,
sikker på at CCS-sekvensering kan produsere 16S leser med en lav frekvens av substitusjon feil, vi neste tenkte at en andel av erstatningen feil innenfor nøyaktig justert leser bør gjenspeile variasjon som kan tilskrives 16S polymorfismer innen en art’ genome12. For eksempel, leser justert til E. coli stamme K-12 substr. MG1655 viste en substitusjon profil, som speilet akkurat som spådd av samkjøre alle syv av 16S-sekvenser som er kjent for å være til stede i denne genome15 (Fig. 2a, c)., Vi var mer i stand til å validere stoichiometry av disse nukleotid erstatninger ved kvantifisering variasjon i relativt justert Illumina WGS leser (Fig. 2b) og viser at en tilsvarende erstatning profil var reproduserbare på tvers av flere sekvensering går (Supplerende Fig. 9)., Tilnærmingene til andre referanse-sekvenser i vår mock fellesskap viste en lignende trend av rikelig innbytter lokalisert til bestemte base posisjoner langs 16S-genet, selv om vi oppmerksom på at signal-til-støy-forholdet økt betraktelig når 16S-genet i spørsmålet hadde færre enn 100 justert leser (Supplerende Fig. 10).

Polymorfismer i E. coli 16S rRNA gen sekvenser. en plassering og frekvens av erstatningene som vises i E., coli stamme K-12 MG1655 V1–V9 amplikoner generert fra våre mock samfunnet og sekvensert på PacBio RS II-plattformen. b plassering og frekvens av innbytter i lyder generert fra genom sekvensering av isolert E. coli stamme K-12 MG1655 på Illumina MiSeq plattform. Forstørret regionene viser respektive posisjoner i justeringen av alle syv 16S genene til stede i E. coli K-12 MG1655 referanse genom. 16S-sekvens fra rrnD operon (**) er brukt som referanse for alle SNP utfasing. c spådd nukleotid substitusjon profil av E., coli K-12 MG1655 basert på å justere de syv 16S gensekvenser til stede i referanse genom. d spådd substitusjon profil av E. coli O157 Sakai basert på å justere de syv 16S gensekvenser til stede i referanse genom. Grå paneler skildre variable regioner definert av brukte primer-bindende områder (Supplerende Tabell 1). Stiplede linjer indikerer den forventede andelen av nukleotid-erstatninger, gitt det er sju 16S genet kopier i hver genom., Kilde data er gitt som en Kilde Data file
Den observasjon at lang-les-sekvensering kan identifisere 16S polymorfismer i samme genom har viktige implikasjoner. For det første, det viser at det ikke er gyldig grunn til å anta at høy gjennomstrømning sekvens leser ulik ved ett eller noen nukleotider representerer en distinkt taxa6,16. Innenfor en enkelt genom, to eller flere 16S sekvenser kan være identiske, mens andre kan være unik., Tilsvarende, noen homologe 16S loci kan beholde samme rekkefølge mellom to nært beslektede stammer, mens andre kan ha skilte seg på en eller noen nukleotid posisjoner. I denne sammenheng bør samfunnet-nivå eller taksonomisk tolkning av 16S data ideelt sett bør konto for det faktum at den relative overflod av 16S sekvenser fremkommer fra veldig nært knyttet taksa vil reflektere en lineær kombinasjon av (i) hvor ofte hver unik sekvens er representert over hele genomer og (ii) den relative overflod av genomet for hver taxon.,
Andre, selv om intragenomic 16S sekvens variasjon kompliserer samfunnet-nivå-analyse, det har også potensial til å øke kraften av 16S-genet til å diskriminere mellom nært beslektede taksa, fordi den gjør det mulig for sekvens-basert forhold til å utvide med flere forskjellige loci. For eksempel, tilstrekkelig nukleotid variasjon eksisterer for å skille E. coli stamme K-12 MG1655 fra enterohemorrhagic belastning O157 Sakai (Fig. 2c, d)., Dermed kan vi argumentere for at, når hensiktsmessig redegjort for, flere polymorfe 16S kopier er ikke en ulempe å bli oversett, men i stedet vil aktivere 16S-genet for å bli brukt i sil-nivå microbiome analyse. Vi ser også at strømmen av intragenomic 16S sekvens variasjon til å diskriminere nært knyttet taksa er sannsynlig å avta når delvis 16S sekvenser er brukt. For eksempel, SNPs skille de E. coli-stammer K-12 MG1655 (Fig. 2c) fra O157 Sakai (Fig. 2d) er funnet i variable regioner V1, V2, V6, og V9.,
16S polymorfismer kan løses i vivo
Microbiome samfunn er ofte komplekse, eksisterende i ulike biokjemiske miljøer (f.eks., avføring, spytt, slim, etc.) og inneholder mange hundre unike taksa som relative overflod spenner over et bredt dynamisk område. Denne kompleksiteten er ikke godt representert i enten silico eller mock samfunnet eksperimenter. Vi har derfor utført en ekstra eksperiment for å vise at sekvensering av hele 16S-genet, mens regnskap for intragenomic 16S SNPs kan løse nært knyttet bakteriell taksa in vivo.,
Vi har gjennomført PacBio CCS-sekvensering av V1–V9-regionen for fire menneskelig avføring prøver fra friske voksne frivillige. Til sammenligning har vi sekvensert i V1–V3-regionen ved hjelp av Illumina MiSeq, og for å gi en standard for arter taksonomisk nivå kvantifisering, har vi utført metagenomic WGS (mWGS) sekvensering ved hjelp av Illumina NextSeq. For å vurdere i hvilken grad hver av disse sekvensering tilnærminger kan løse nært knyttet taksa, vi fokusert på slekten Bacteroides., I tillegg til å være rike i den menneskelige tarmen, denne slekten er svært ulike, inneholder flere arter som kan utøve både gode og dårlige virkninger på menneskelig health17. Det har også vært brukt som modell taxon for å demonstrere nytten av 16S genet for høy oppløsning taksonomisk analysis18.
Når vi beregnet Bacteroides overflod på slekten nivå, V1–V9-sekvensering og V1–V3-sekvensering produsert sammenlignbare resultater., Begge tilnærminger identifisert to personer med lave Bacteroides relative overflod (~10-25%) og to individer med høy Bacteroides relative overflod (~40-60%; Fig. 3a). Imidlertid, arter-nivå kvantifisering via mWGS sekvensering åpenbart langt større mangfold, med en annen Bacteroides arter som er dominerende i tarmen av hver enkelt (Fig. 3b og Supplerende Data 1). Når clustering OTUs på 99% identitet, både V1–V9 og V1–V3-sekvensering var i stand til å reflektere denne arten-nivå variasjon (Fig., 3b), med det unntak at V1–V3-sekvensering ikke oppdage Bacteroides cystoids, som var rikelig i en av de fire menneskelige tarmen microbiome prøver. Basert på disse resultatene kan vi konkludere med at, når det brukes i forbindelse med en passende identitet terskel (f.eks., 99%), OTU-baserte tilnærminger har potensial til å løse arter-nivå mangfold observert i den menneskelige tarmen. Vi videre oppmerksom på at selv om full-lengde sekvensering av 16S kan være optimal for arter-nivå analyse, svært informativ variable regioner (f.eks., V1–V3) kan også være tilstrekkelige for dette formålet.,

å Oppdage Bacteroides i menneskelig avføring prøver. en relativ overflod av slekten Bacteroides i fire menneskelig avføring prøver kvantifisert ved hjelp av enten V1–V9 amplikoner (x-aksen) eller V1–V3 amplikoner (y-aksen). b Den relative overflod av Bacteroides arter i de samme fire prøver. Arter overflod ble kvantifisert fra mWGS sekvensering eller fra V1–V3/V1–V9 OTUs generert på 99% identitet., Overflod er vist for de mest tallrike artene som kvantifisert ved mWGS (for tallrikhetsestimater av alle Bacteroides arter registrert ved hver plattform, se Utfyllende Tabell 5). c Nukleotid substitusjon profiler blir generert ved å justere alle V1–V9-amplikon sekvenser som er tilordnet enkelt OTU identifisert som Bacteroides vulgatus. Profilene er vist for de to krakk prøver med høy B. vulgatus relative overflod (IronHorse og Scott). d Nukleotid substitusjon profiler spådd fra referanse genomet til to forskjellige B. vulgatus stammer ATCC 848239 og mpk40., I både c-og d -, nukleotid-erstatninger ble identifisert i forhold til en enkelt referanse 16S genet for B. vulgatus ATCC 8482 (NCBI Genet ID 5304800). Grå paneler skildre variable regioner definert av brukte primer-bindende områder (Supplerende Tabell 1). Stiplede linjer indikerer den forventede andelen av nukleotid-erstatninger, gitt det er sju 16S genet kopier i hver genom., Kilde data er gitt som en Kilde Data file
– >
Tar nytte av det faktum at Bacteroides vulgatus var til stede på høy relativ overflod i to av våre menneskelige tarmen microbiome prøver vi neste spurte om intragenomic variasjon mellom 16S genet kopier kan bli oppdaget i vivo. Vi har justert alle full-lengde sekvens klassifisert som tilhører vårt B. vulgatus V1–V9 OTUs (Fig. 3b og Supplerende Data 1) til en enkelt representant B. vulgatus 16S genet rekkefølge. Vi så sammenlignet den resulterende nukleotid substitusjon profiler (Fig., 3c) med profiler spådd fra to referanse-genomet til stede i NCBI RefSeq database19 (Fig. 3d).
flertallet av nukleotid variasjon til stede i vår in vivo generert B. vulgatus OTU reflektert sant variasjon som kan tilskrives intragenomic polymorfismer. I kontrast, variasjon sannsynlig på grunn av sekvensering feil dukket opp lavt og godt under minimal ~14% frekvens som forventes hvis det var en enkelt B. vulgatus belastningen i hver prøve med syv 16S genet eksemplarer i sin genom (Fig. 3c, stiplede linjer).
Selv om vi ikke kjenner den sanne antall B., vulgatus stammer til stede i alle in-vivo eksempel, det var bemerkelsesverdig at både nukleotid substitusjon profiler bar nærmere likhet med stamme ATCC 8482 enn mpk. Variasjon fantes også på spesifikke loci som potensielt kan indikere meningsfulle forskjeller mellom in vivo og ATCC 8482 referanse genomer. For eksempel kan en enkelt polymorphism ble oppdaget i V5-regionen i ATCC 8482, som var til stede i tre 16S kopier (43%). I første in-vivo eksempel (Scott) dette polymorphism var til stede i 84% av lyder, mens i den andre (IronHorse) det var til stede i 69% av lyder., Disse tallene tilsvarer tett til tallene forventes hvis en polymorphism var til stede seks og fem av syv 16S gener, henholdsvis.
I konklusjonen, viser vi at full-lengde 16S sekvensering av den menneskelige tarmen microbiome kan nøyaktig løse enkelt-nukleotid erstatninger som gjenspeiler intragenomic variasjon mellom 16S genet kopier. Tilstedeværelse av en slik variasjon indikerer at 16S sekvenser må være gruppert å reflektere meningsfylt taksonomisk enheter., Ved hjelp av OTUs gruppert på 99% identitet, viser vi at full-lengde 16S har potensial til å gi arter og til og med sil-nivå taksonomisk oppløsning. Analyse av mikrobielle samfunn på disse taksonomisk nivå lover å gi et helt annet perspektiv til en by av slekten-nivå tallrikhetsestimater.
Intragenomic 16S polymorfismer er svært utbredt
etter å Ha vist at det er mulig å løse intragenomic kopi varianter in vivo, vi neste søkt å etablere den grad slike kopier varianter vises i taksa som ofte finnes i den menneskelige tarmen microbiome., Vi har videre søkt å fastslå om slike profiler kan også rutinemessig brukes for å skille mellom stammer av samme art.
Vi dyrket 381 taksa fra tarmen microbiome av de friske personene som er avbildet i Fig. 3, så vel som fra andre personer å delta i samme originale study20 (Supplerende Data 2). Vi senere utført full-lengde 16S gen sekvenser på isolerer og justert sekvensert leser å identifisere nukleotid innbytter karakteristisk for intragenomic 16S genet kopi varianter.,
Taksonomisk klassifisering av isolater identifisert 58 antatte arter (Supplerende Data 2), mens clustering en enkelt representant sekvens for hver isolere på 99% likhet resulterte i 61 OTUs (med mellom 1 og 73 isolater som er tilordnet til hver OTU). I sum, 349 av 381 sekvensert isolater (54 av 61 OTUs) hadde ett eller flere SNP, som indikerer tilstedeværelsen av 16S genet polymorfismer, og 205 unike SNP-profiler ble identifisert ved regnskapsføring av potensielle sekvensering feil (Fig. 4a og Supplerende Data 2).

Intragenomic 16S genet polymorfismer i menneskelig gut microbiome isolater. en Plassering av SNPs til stede i 16S gener av individuelt kultivert bakterielle isolater. SNP steder ble identifisert gjennom innfasing full-lengde 16S gen sekvenser generert for hver enkelt isolere. X-aksen angir posisjon langs 16S-genet. Y-aksen angir enkelte isolater gruppert basert på deres anslåtte fylogeni. Mørk blå regionen angir plasseringen av en polymorphism., For klarhet, maksimalt fem isolater som hører til samme art er vist. For detaljer om nukleotid substitusjon profiler for alle sekvensert isolater, se Utfyllende Data 2. b–d Eksempler på nukleotid substitusjon profiler som viser sil-nivå forskjeller mellom isolater identifisert som tilhørende tre bakteriearter: b Shigella flexneri; c Bifidobacterium longum; d Collinsella aerofaciens. For hver art, to isolere nukleotid substitusjon profilene er vist, men flere eksempler kan være funnet i Supplerende Data 2., Isolatene ble identifisert som tilhører samme art hvis deres representant sekvenser ble tildelt samme OTU når clustering på 99% sekvens identitet. Taksonomisk identifikasjon ble utført ved hjelp SPRENG for å justere representant sekvenser til NCBI 16S BLAST databasen (se Metoder). Grå paneler skildre variable regioner definert av brukte primer-bindende områder (Supplerende Tabell 1). Stiplede linjer indikerer den forventede andelen av nukleotid-erstatninger, gitt antall 16S genet kopier spådd for hver genom., Kilde data er gitt som en Kilde Data file
først og fremst å sammenligne SNP-profiler for isolater som er tilordnet den samme OTU ofte avdekket forskjeller i frekvensen av SNPs som ble tyder på forskjeller i intragenomic 16S genet kopier mellom nært beslektede taksa. Eksempler på ulike substitusjon profilene er vist for tre taksa (Fig. 4b–d), som er betont av sil-nivå variasjon sammenlignbart med det vi viste i prinsippet for E. coli (Fig. 2b).,
I konklusjonen, viser vi at mange av de culturable medlemmer av den menneskelige tarmen microbiome ofte har 16S genet polymorfismer, som, når det redegjort for, har potensial til å løse stammer av samme art.