pełny Gen 16S zapewnia lepszą rozdzielczość taksonomiczną
Gen ~1500 bp 16S rRNA obejmuje dziewięć zmiennych regionów przeplatanych przez wysoce zachowaną sekwencję 16S (rys. 1a). Sekwencjonowanie całego genu zostało pierwotnie osiągnięte przez sekwencjonowanie Sangera., Wymagało to klonowania genów, generowania i montażu od dwóch do trzech odczytów na klon i wytwarzania ograniczonej głębokości pobierania próbek przy wysokich kosztach i wysiłku. Obecnie jednak zdecydowana większość badań sekwencjonuje tylko część genu, ponieważ szeroko stosowana Platforma sekwencjonowania Illumina (większa przepustowość, niższy koszt, mniejszy wysiłek w porównaniu z Sangerem) wytwarza krótkie sekwencje (≤300 baz)., Różne subregiony genu są zatem ukierunkowane, począwszy od pojedynczych zmiennych regionów, takich jak V4 lub V6, do trzech zmiennych regionów, takich jak V1-V3 lub V3-V5 (stosowane w projekcie mikrobiomu ludzkiego w połączeniu z platformą sekwencjonującą 4549).
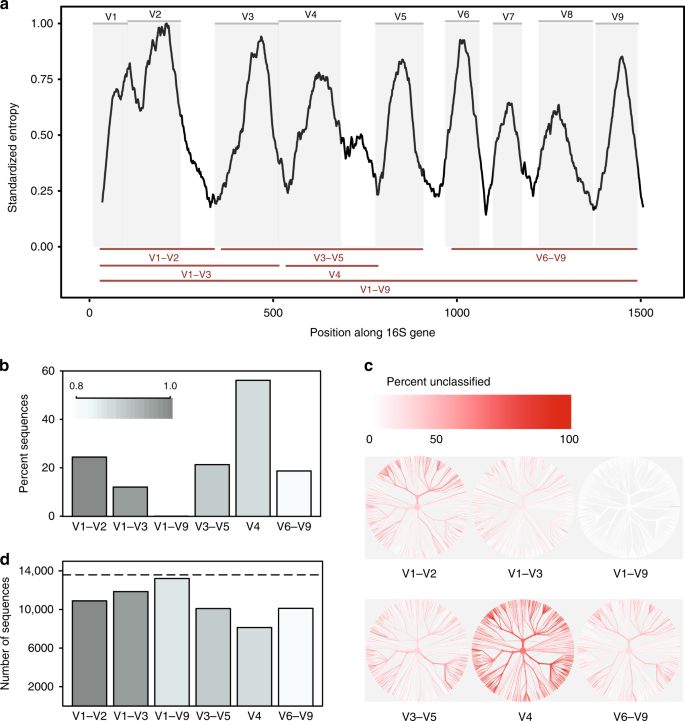
In-Silico porównanie regionów zmiennych 16S rRNA. Entropia Shannona w genie 16S oparta na wyrównaniu pojedynczej sekwencji reprezentatywnej dla każdego znanego gatunku występującego w bazie danych Greengenesa., Sekwencje zostały dopasowane do pojedynczego referencyjnego genu 16S dla Escherichia coli K-12 MG1655 (Gen NCBI ID 947777). Szare panele przedstawiają zmienne regiony zdefiniowane przez powszechnie stosowane miejsca wiązania podkładu (tabela uzupełniająca 1). Regiony zmienne rozważane w tym badaniu są pokazane jako czerwone linie (na dole). b odsetek sekwencji dla każdego regionu zmiennego, którego nie można było zidentyfikować do poziomu gatunku przy klasyfikacji każdej sekwencji w stosunku do referencyjnej bazy danych, z której została ona uzyskana, przy progu ufności wynoszącym 80% (klasyfikator PROW)., drzewa c na podstawie taksonomii sekwencji występujących w bazie in-silico. To samo drzewo jest dostarczane dla każdego regionu zmiennej. Kolor każdej gałęzi odzwierciedla proporcję sekwencji w obrębie każdego kladu, których nie można zidentyfikować do poziomu gatunku. d Liczba otu utworzonych podczas grupowania sekwencji dla każdego regionu zmiennej przy 99% podobieństwie sekwencji. Linia przerywana wskazuje liczbę unikalnych sekwencji (>1% różnych) w oryginalnej bazie danych., Dane źródłowe są dostarczane jako plik danych źródłowych
argumentujemy, że kierowanie podregionów stanowi historyczny kompromis ze względu na ograniczenia technologiczne10. Dziś, zarówno PacBio i Oxford Nanopore sekwencjonowania platformy są zdolne do rutynowo produkujących czyta w ponad 1500 bp i wysokiej przepustowości sekwencjonowanie pełnego genu 16s staje się coraz bardziej powszechne., Dlatego sugerujemy, że uzasadnienie tego kompromisu wymaga ponownego rozpatrzenia i przeprowadziliśmy prosty eksperyment in-silico, aby wykazać przewagę sekwencjonowania 16S o Pełnej długości nad ukierunkowaniem subregionów.
ściągnęliśmy zestaw nie nadmiarowych (tj.> 1% różnych), pełnej długości sekwencji 16S z publicznej bazy danych (Greengenes)., Korzystając z faktu, że znaczna część tych sekwencji zawiera miejsca wiążące primer PCR, przycięliśmy je do generowania amplikonów in-silico dla różnych podregionów, w oparciu o lokalizację primerów PCR powszechnie stosowanych w badaniach mikrobiomu (rys. 1a oraz tabele uzupełniające 1-2)., Zakładając, że każda sekwencja w naszej pobranej bazie danych reprezentuje unikalny gatunek, zastosowaliśmy wspólne podejście klasyfikacyjne (klasyfikator projektu bazy rybosomów (RDP) 11), aby obliczyć częstotliwość, z jaką amplikony in-silico dla każdego podregionu mogą zapewnić dokładną klasyfikację taksonomiczną na poziomie gatunku (używając oryginalnej bazy danych jako odniesienia). W drugim eksperymencie zebraliśmy również nasze amplikony in-silico, aby wygenerować OTUs przy różnych, powszechnie używanych, progach podobieństwa sekwencji(97%, 98%, 99%).,
odkryliśmy, że podregiony różniły się znacznie w zakresie, w jakim mogły śmiało rozróżniać sekwencje 16S o Pełnej długości używane do reprezentowania gatunków (rys. 1b). Region V4 wypadł najgorzej, z 56% amplikonów in-silico nie zgadzało się z sekwencją ich pochodzenia na tym poziomie taksonomicznym. Natomiast, gdy użyto sekwencji pełnej długości ze wszystkimi zmiennymi regionami, można było sklasyfikować prawie wszystkie sekwencje jako właściwe gatunki(rys. 1a)., Zmiana baz danych i progów ufności w klasyfikacji wpłynęła na odsetek amplikonów in-silico, które można było dokładnie dopasować, ale nie wpłynęła na panujące trendy (rys. 1a, b).
Po drugie, różne podregiony wykazały stronniczość w taksonach bakterii, które były w stanie zidentyfikować (rys. 1c). Na przykład region V1–V2 wypadł słabo w klasyfikacji sekwencji należących do Phylum Proteobacteria, podczas gdy region V3-V5 wypadł słabo w klasyfikacji sekwencji należących do Phylum Actinobacteria(dodatkowe rys. 2)., Podobne tendencje zaobserwowano na poziomie rodzaju dla taksonów o potencjalnym znaczeniu medycznym. Chociaż pełny region V1-V9 konsekwentnie dawał najlepsze wyniki, region V6-V9 był szczególnie najlepszym podregionem do klasyfikacji sekwencji należących do rodzajów Clostridium i Staphylococcus, region V3-V5 dawał dobre wyniki dla Klebsiella, a region V1-V3 dawał dobre wyniki dla Escherichia / Shigella (dodatkowe rys. 2 oraz dane źródłowe).
wreszcie wybór podregionu dramatycznie wpłynął na liczbę otu utworzonych podczas grupowania amplikonów in-silico w celu utworzenia otu., Podczas klastrowania przy 99% tożsamości sekwencji, wszystkie podregiony nie udało się odtworzyć liczby odrębnych sekwencji obecnych w oryginalnej bazie danych; jednak Region V4 ponownie wypadł najgorzej (rys. 1d). W szczególności względna liczba otu wyprodukowanych przez każdy podregion nie była spójna przy różnych progach tożsamości (97%, 98%, 99%, dodatkowe rys. 3), wskazując, że zachowanie algorytmów grupowania może być trudne do przewidzenia, gdy ilość informacji zawartych w uporządkowanym regionie jest wysoce zmienna.,
podsumowując, ukierunkowanie podregionów stanowi historyczny kompromis, który był wystarczający do identyfikacji taksonów na poziomie rodzaju lub powyżej. Jednak nasz prosty eksperyment in-silico pokazuje, że nie jest słuszne zakładanie, że coraz dokładniejsze grupowanie tych podregionów spowoduje poprawę rozdzielczości taksonomicznej koniecznej do odzwierciedlenia gatunków. Chociaż niektóre podregiony (np. V1–V3) zapewniają rozsądne przybliżenie różnorodności 16S, większość z nich nie wychwytuje wystarczającej zmienności sekwencji, aby rozróżnić blisko spokrewnione taksony., Zauważamy również, że dyskryminujące polimorfizmy mogą być ograniczone do określonych zmiennych regionów; w ten sposób niektóre podregiony będą lepiej przystosowane do rozróżniania blisko spokrewnionych członków niektórych taksonów.
warianty kopii genu 16S odzwierciedlają zmienność poziomu szczepu
grupowanie sekwencji 16S w OTUs w przeszłości służyło dwóm celom. Po pierwsze, usunięto drobne artefaktualne warianty sekwencji z powodu amplifikacji PCR i błędów sekwencjonowania podczas zwijania sekwencji w grupy. Po drugie, zawalił legalne warianty sekwencji, które istnieją między blisko spokrewnionymi taksonami bakteryjnymi., Chociaż ta ostatnia może nie zawsze być pożądana, nie można odróżnić taksonów bakteryjnych, których sekwencje 16S różnią się w tempie niższym niż błąd napotkany na danej platformie sekwencyjnej.
Ostatnio postępy w CCS znacznie poprawiły wskaźniki błędów platform sekwencjonowania o długim odczycie. Jednocześnie, metody obliczeniowe umożliwiły rozróżnienie między uzasadnioną a artefaktualną zmiennością sekwencji., Te technologiczne i metodologiczne postępy oznaczają, że naukowcy mają obecnie potencjał do wykonywania sekwencjonowania o wysokiej przepustowości, które mogą dokładnie wykryć warianty pojedynczego nukleotydu w całym Genie 16S.
chociaż kuszące jest założenie, że warianty jednonukleotydowe mogą reprezentować odrębne, blisko spokrewnione taksony,przestrzegamy przed tą zbyt uproszczoną interpretacją ze względu na fakt,że wiele genomów bakteryjnych zawiera wiele polimorficznych kopii genów 16S 12, 13, 14., Przeprowadziliśmy sekwencjonowanie PacBio CCS 36 gatunków bakterii mock community (dodatkowa Tabela 3 i dodatkowa rys. 4) wykazanie (i), że sekwencja 16S wielu bakterii zmienia się między operonami w obrębie tego samego genomu oraz (ii), że sekwencjonowanie o wysokiej przepustowości jest wystarczająco dokładne, aby rozwiązać te różnice wewnątrzgenomiczne.
dopasowaliśmy sekwencje PacBio o Pełnej długości 16S do referencyjnej bazy danych zawierającej pojedynczą reprezentatywną sekwencję 16S dla każdego członka naszej mock community i wykorzystaliśmy statystyki wyrównania do oceny dokładności tego podejścia do sekwencjonowania., Porównanie liczby przejść użytych do wygenerowania CCS z występowaniem podstawień, wstawień i delecji jednonukleotydowych wykazało, że dziesięć przejść może zminimalizować te połączone błędy do minimalnej częstotliwości < 1.0% (chociaż zauważalne było, że minimalny możliwy do osiągnięcia błąd różnił się między sekwencjonowaniem przebiegów; dodatkowe rys. 5). Jednak zaobserwowaliśmy zbieżność błędów delecji z homopolimerem lokalizacji w naszych sekwencjach referencyjnych (rys., 6), który nie był specyficzny dla nukleotydów i był pogarszany długością sekwencjonowanego homopolimeru (rys. 7). Następnie zwalidowaliśmy delecje w obrębie genu Escherichia coli 16S za pomocą sekwencjonowania Illumina whole genome shotgun (WGS), co wykazało, że tylko jedna z delecji występujących w sekwencjach PacBio była prawdziwa (dodatkowa rys. 8).,
przekonani, że sekwencjonowanie CCS może wytwarzać odczyty 16S z niską częstotliwością błędów substytucji, stwierdziliśmy następnie, że część błędów substytucji w dokładnie wyrównanych odczytach powinna odzwierciedlać zmienność przypisaną polimorfizmom 16S w obrębie genomu gatunkowego12. Na przykład odczyty wyrównane do szczepu E. coli K-12 podst. MG1655 wykazał profil substytucji, który odzwierciedlał dokładnie to, co przewidywano, wyrównując wszystkie siedem sekwencji 16S znanych z obecności w tym genomie15 (rys. 2a, c)., Byliśmy również w stanie zweryfikować stechiometrię tych podstawień nukleotydów poprzez ilościowe określenie zmienności w porównywalnie wyrównanych odczytach Illumina WGS (rys. 2b) i wykazać, że podobny profil substytucji był odtwarzalny w wielu sekwencjach (rys. 9)., Wyrównania do innych sekwencji referencyjnych w naszej mock community wykazały podobny trend obfitych podstawień zlokalizowanych w określonych pozycjach bazowych wzdłuż genu 16S, chociaż zauważamy, że stosunek sygnału do szumu znacznie wzrósł, gdy gen 16S, o którym mowa, miał mniej niż 100 wyrównanych odczytów (dodatkowe rys. 10).

polimorfizmy w sekwencjach genów rRNA E. coli 16S. a pozycja i częstotliwość zastępstw pojawiających się w E., szczep coli K-12 MG1655 V1-amplikony V9 generowane z naszej mock community I sekwencjonowane na platformie PacBio RS II. b pozycja i częstotliwość podstawień w odczytach generowanych z sekwencjonowania genomowego izolowanego szczepu E. coli K-12 MG1655 na platformie Illumina MiSeq. Powiększone regiony pokazują odpowiednie pozycje w wyrównaniu wszystkich siedmiu genów 16S obecnych w genomie referencyjnym E. coli K-12 MG1655. Sekwencja 16S z rrnd operon (**) jest używana jako odniesienie dla wszystkich faz SNP. c przewidywany profil substytucji nukleotydów E., coli K-12 MG1655 w oparciu o dostosowanie siedmiu sekwencji genów 16S obecnych w genomie referencyjnym. d przewidywany profil substytucji E. coli O157 Sakai oparty na wyrównaniu siedmiu sekwencji genów 16S obecnych w genomie referencyjnym. Szare panele przedstawiają zmienne regiony zdefiniowane przez powszechnie stosowane miejsca wiązania podkładu (tabela uzupełniająca 1). Linie przerywane wskazują oczekiwany udział podstawień nukleotydów, biorąc pod uwagę, że w obrębie każdego genomu znajduje się siedem kopii genu 16S., Dane źródłowe są dostarczane jako plik danych źródłowych
obserwacja, że długo czytane sekwencjonowanie może zidentyfikować polimorfizmy 16S w obrębie tego samego genomu ma istotne implikacje. Po pierwsze, dowodzi, że nie jest słuszne zakładanie, że sekwencja o dużej przepustowości różniąca się jednym lub kilkoma nukleotydami reprezentuje odrębny taksa6, 16. W obrębie jednego genomu dwie lub więcej sekwencji 16S mogą być identyczne, podczas gdy inne mogą być unikalne., Odpowiednio, niektóre homologiczne loci 16S mogą zachować identyczną sekwencję pomiędzy dwoma blisko spokrewnionymi szczepami, podczas gdy inne mogą się różnić w jednej lub kilku pozycjach nukleotydów. W tym kontekście każda interpretacja na poziomie wspólnotowym lub taksonomiczna danych 16S powinna idealnie uwzględniać fakt, że względna obfitość sekwencji 16S pochodzących z bardzo blisko spokrewnionych taksonów będzie odzwierciedlać kombinację liniową (i) częstotliwości, z jaką każda unikalna sekwencja jest reprezentowana w genomach oraz (ii) względnej obfitości genomów dla każdego taksonu.,
Po drugie, chociaż zmienność sekwencji 16S komplikuje analizę na poziomie Wspólnoty, ma również potencjał zwiększenia mocy genu 16S do rozróżniania między blisko spokrewnionymi taksonami, ponieważ umożliwia porównanie oparte na sekwencji rozciąganie się na wiele rozbieżnych loci. Na przykład istnieje wystarczająca zmienność nukleotydów, aby odróżnić szczep E. coli K-12 MG1655 od szczepu jelitowo-krwotocznego O157 Sakai (rys. 2c, d)., W związku z tym argumentujemy, że po odpowiednim uwzględnieniu wielokrotnych polimorficznych kopii 16S nie jest niewygodą, którą należy przeoczyć, a raczej umożliwi wykorzystanie genu 16S w analizie mikrobiomu na poziomie szczepu. Zauważamy również, że moc intragenomicznej zmienności sekwencji 16S do rozróżniania blisko spokrewnionych taksonów prawdopodobnie zmniejszy się, gdy używane są częściowe sekwencje 16S. Na przykład, SNP odróżniające szczepy E. coli K-12 MG1655 (rys. 2C) z O157 Sakai (rys. 2d) znajdują się w zmiennych regionach V1, V2, V6 i V9.,
polimorfizmy 16S można rozwiązać in vivo
społeczności mikrobiomów są często złożone, istniejące w różnych środowiskach biochemicznych (np. stolec, ślina, plwocina itp.) i zawierający wiele setek unikalnych taksonów, których względna liczebność obejmuje szeroki zakres dynamiczny. Ta złożoność nie jest dobrze reprezentowana w eksperymentach in-silico lub mock community. Dlatego przeprowadziliśmy dodatkowy eksperyment w celu wykazania, że sekwencjonowanie pełnego genu 16S przy uwzględnieniu intragenomic 16s SNPs może rozwiązać blisko spokrewnione taksony bakteryjne in vivo.,
przeprowadziliśmy sekwencjonowanie PacBio CCS regionu V1–V9 dla czterech próbek kału ludzkiego pobranych od zdrowych dorosłych ochotników. Dla porównania zsekwencjonowaliśmy region V1–V3 za pomocą Illumina MiSeq i, aby zapewnić punkt odniesienia dla kwantyfikacji taksonomicznej na poziomie gatunków, przeprowadziliśmy sekwencjonowanie metagenomiczne WGS (mWGS) przy użyciu Illumina NextSeq. Aby ocenić, w jakim stopniu każde z tych podejść sekwencjonowania może rozwiązać blisko spokrewnione taksony, skupiliśmy się na rodzaju Bacteroides., Oprócz obfitości w ludzkie jelita, rodzaj ten jest bardzo zróżnicowany i zawiera wiele gatunków, które mogą wywierać zarówno dobry, jak i zły wpływ na zdrowie ludzkie17. Wcześniej był on również wykorzystywany jako model taksonu do wykazania przydatności genu 16S do analizy taksonomicznej o wysokiej rozdzielczości18.
Kiedy obliczyliśmy liczebność Bacteroides na poziomie rodzaju, sekwencjonowanie V1–V9 i V1–V3 dało porównywalne wyniki., Oba podejścia zidentyfikowały dwa osobniki o niskiej względnej obfitości Bacteroides (~10-25%) i dwa osobniki o wysokiej względnej obfitości Bacteroides (~40-60%; rys. 3a). Jednak ocena ilościowa na poziomie gatunku za pomocą sekwencjonowania mWGS wykazała znacznie większą różnorodność, z różnymi gatunkami Bacteroides dominującymi w jelitach każdego osobnika(rys. 3b oraz dane uzupełniające 1). Podczas grupowania OTUs przy 99% tożsamości, zarówno sekwencjonowanie V1–V9, jak i V1–V3 były w stanie odzwierciedlić tę zmienność na poziomie gatunku (rys., 3b), z zauważalnym wyjątkiem, że sekwencjonowanie V1–V3 nie wykryło Bacteroides intestinalis, które było obfite w jednej z czterech próbek mikrobiomu jelitowego człowieka. Na podstawie tych wyników wnioskujemy, że w połączeniu z odpowiednim progiem tożsamości (np. 99%), podejścia oparte na OTU mają potencjał do rozwiązania różnorodności na poziomie gatunkowym obserwowanej w jelitach człowieka. Ponadto zauważamy, że chociaż sekwencjonowanie pełnej długości 16S może być optymalne dla analizy na poziomie gatunku, wysoce informatywne zmienne regiony (np. V1–V3) mogą być również odpowiednie do tego celu.,

wykrywanie Bacteroides w ludzkich próbkach kału. a względna liczebność rodzaju Bacteroides w czterech próbkach kału ludzkiego określona ilościowo za pomocą amplikonów V1–V9 (oś x) lub V1-V3 (oś y). b względna liczebność gatunków Bacteroides w tych samych czterech próbkach. Liczebność gatunku określano ilościowo na podstawie sekwencjonowania mWGS lub na podstawie OTUs V1–V3/V1–V9 wygenerowanych przy 99% tożsamości., Liczebność jest wykazywana w odniesieniu do najbardziej obfitych gatunków zgodnie z kwantyfikacją mWGS (szacunki liczebności wszystkich gatunków Bacteroides wykrytych przez poszczególne platformy, patrz tabela dodatkowa 5). c profile substytucji nukleotydów generowane przez wyrównanie wszystkich sekwencji amplikonowych V1–V9 przypisanych do pojedynczego OTU zidentyfikowanego jako Bacteroides vulgatus. Profile są pokazane dla dwóch próbek kału o wysokiej względnej liczebności B. vulgatus(IronHorse i Scott). D profile substytucji nukleotydów przewidywane na podstawie genomów referencyjnych dwóch różnych szczepów B. vulgatus ATCC 848239 i mpk40., Zarówno w c, jak i d, podstawienia nukleotydów zidentyfikowano w stosunku do pojedynczego referencyjnego genu 16S dla B. vulgatus ATCC 8482 (Gen NCBI ID 5304800). Szare panele przedstawiają zmienne regiony zdefiniowane przez powszechnie stosowane miejsca wiązania podkładu (tabela uzupełniająca 1). Linie przerywane wskazują oczekiwany udział podstawień nukleotydów, biorąc pod uwagę, że w obrębie każdego genomu znajduje się siedem kopii genu 16S., Dane źródłowe są dostarczane jako plik danych źródłowych
korzystając z faktu, że Bacteroides vulgatus był obecny przy wysokiej względnej obfitości w dwóch naszych próbkach mikrobiomu jelitowego człowieka, zapytaliśmy następnie, czy zmienność wewnątrzgenomowa między kopiami genu 16S może być wykryta in vivo. Wyrównaliśmy wszystkie sekwencje pełnej długości sklasyfikowane jako należące do naszego B. vulgatus V1–V9 OTUs (rys. 3b i dane uzupełniające 1) do pojedynczej sekwencji genu B. vulgatus 16S. Następnie porównaliśmy uzyskane profile substytucji nukleotydów (rys., 3c) z profilami przewidzianymi dla dwóch genomów referencyjnych obecnych w bazie danych NCBI Refseq19 (rys. 3d).
większość wariacji nukleotydów obecnych w naszym In vivo generowanych B. vulgatus otu odzwierciedlała prawdziwą zmienność przypisywaną polimorfizmom intragenomicznym. W przeciwieństwie do tego, zmienność prawdopodobnie spowodowana błędami sekwencjonowania pojawiła się niska i znacznie poniżej minimalnej ~14% częstotliwości, której można by się spodziewać, gdyby w każdej próbce znajdował się pojedynczy szczep B. vulgatus z siedmioma kopiami genu 16S w genomie (rys. 3C, linie przerywane).
chociaż nie znaliśmy prawdziwej liczby B., szczepy vulgatus obecne w każdej próbce in vivo, zauważalne było, że oba profile substytucji nukleotydów miały bliższe podobieństwo do szczepu ATCC 8482 niż mpk. Zmienność istniała również w specyficznych loci, które potencjalnie mogą wskazywać znaczące różnice między In vivo i ATCC 8482 genomów referencyjnych. Na przykład pojedynczy polimorfizm wykryto w regionie V5 ATCC 8482, który był obecny w trzech egzemplarzach 16S (43%). W pierwszej próbce in vivo (Scott) polimorfizm ten był obecny w 84% odsłon, podczas gdy w drugiej (IronHorse) był obecny w 69% odsłon., Liczby te odpowiadają ściśle liczbom oczekiwanym w przypadku obecności polimorfizmu odpowiednio sześciu i pięciu z siedmiu genów 16S.
podsumowując, pokazujemy, że sekwencjonowanie 16S w pełnej długości ludzkiego mikrobiomu jelitowego może dokładnie rozwiązać podstawienia jednonukleotydowe, które odzwierciedlają zmienność wewnątrzgenomiczną między kopiami genu 16S. Obecność takiej zmienności wskazuje, że sekwencje 16S muszą być grupowane w celu odzwierciedlenia znaczących jednostek taksonomicznych., Wykorzystując OTUs skupiony w 99% tożsamości, pokazujemy, że pełnowymiarowe 16 ma potencjał, aby zapewnić rozdzielczość taksonomiczną na poziomie gatunku, a nawet szczepu. Analiza zbiorowisk drobnoustrojów na tych poziomach taksonomicznych obiecuje zapewnić zupełnie inną perspektywę niż ta zapewniona przez szacunki liczebności na poziomie rodzaju.
Intragenomic 16s polimorfizmy są bardzo rozpowszechnione
po wykazaniu, że możliwe jest rozwiązanie intragenomic kopii warianty in vivo, my next starał się ustalić zakres, w jakim takie warianty kopii pojawiają się w taksonach powszechnie występujących w ludzkim mikrobiomie jelitowym., Ponadto staraliśmy się ustalić, czy takie profile mogą być rutynowo wykorzystywane do rozróżniania szczepów tego samego gatunku.
wyhodowaliśmy 381 taksonów z mikrobiomu jelitowego zdrowych osobników przedstawionych na Rys. 3, jak również od innych osób uczestniczących w tym samym pierwotnym badaniu20 (dane uzupełniające 2). Następnie przeprowadziliśmy sekwencjonowanie genu 16S na izolatach i wyrównaliśmy sekwencjonowane odczyty, aby zidentyfikować podstawienia nukleotydów charakterystyczne dla wewnątrzgenomowych wariantów kopii genu 16S.,
w klasyfikacji taksonomicznej izolatów zidentyfikowano 58 domniemanych gatunków (dane uzupełniające 2), natomiast grupowanie pojedynczej sekwencji reprezentatywnej dla każdego izolatu przy 99% podobieństwie dało 61 OTUs (z od 1 do 73 izolatów przypisanych do każdego OTU). W sumie, 349 z 381 sekwencjonowanych izolatów (54 Z 61 OTUs) miał JEDEN lub więcej SNP, co wskazuje na obecność polimorfizmów genu 16S, i 205 unikalnych profili SNP zidentyfikowano przy uwzględnieniu potencjalnego błędu sekwencjonowania (rys. 4a oraz dane uzupełniające 2).

Wewnątrzgenomiczne polimorfizmy genu 16S w izolatach mikrobiomu jelitowego człowieka. lokalizacja SNP obecnych w genach 16S indywidualnie hodowanych izolatów bakteryjnych. Lokalizacje SNP zostały zidentyfikowane poprzez fazowanie sekwencji genów 16S o Pełnej długości generowanych dla każdego izolatu. Oś X oznacza pozycję wzdłuż genu 16S. Oś Y oznacza poszczególne Izolaty grupowane na podstawie ich wnioskowanej filogenezy. Ciemnoniebieski obszar wskazuje na położenie polimorfizmu., Dla jasności przedstawiono maksymalnie pięć izolatów należących do tego samego gatunku. Szczegółowe informacje na temat profili substytucji nukleotydów dla wszystkich sekwencjonowanych izolatów znajdują się w danych uzupełniających 2. B-D przykłady profili substytucji nukleotydów wykazujących różnice w poziomie szczepów między izolatami zidentyfikowanymi jako należące do trzech gatunków bakterii: B Shigella flexneri; C Bifidobacterium longum; D Collinsella aerofaciens. Dla każdego gatunku przedstawiono dwa izolowane profile substytucji nukleotydów; jednak dodatkowe przykłady można znaleźć w danych uzupełniających 2., Izolaty zostały zidentyfikowane jako należące do tego samego gatunku, jeżeli ich sekwencje reprezentatywne zostały przypisane do tego samego OTU podczas grupowania przy 99% tożsamości sekwencji. Identyfikacja taksonomiczna została przeprowadzona za pomocą BLAST w celu dostosowania sekwencji reprezentatywnych do bazy danych NCBI 16S BLAST (patrz metody). Szare panele przedstawiają zmienne regiony zdefiniowane przez powszechnie stosowane miejsca wiązania podkładu (tabela uzupełniająca 1). Linie przerywane wskazują oczekiwany udział podstawień nukleotydów, biorąc pod uwagę liczbę kopii genu 16S przewidywanych dla każdego genomu., Dane źródłowe są dostarczane jako plik danych źródłowych
porównywanie profili SNP dla izolatów przypisanych do tego samego OTU często ujawniało różnice w częstości występowania SNP, które sugerowały różnice w wewnątrzgenomicznych kopiach genu 16S między blisko spokrewnionymi taksonami. Przykłady różnych profili substytucji przedstawiono dla trzech taksonów (rys. 4b-d), które sugerują zmianę poziomu szczepu porównywalną z tą, którą wykazaliśmy w zasadzie dla E. coli (rys. 2b).,
podsumowując, pokazujemy, że wielu hodowanych członków ludzkiego mikrobiomu jelitowego często posiada polimorfizmy genu 16S, które, gdy są prawidłowo rozliczone, mają potencjał do rozwiązania szczepów tego samego gatunku.