sekwencjonowanie RNA (Wang 2009) szybko zastępuje mikromacierze ekspresji genów w wielu laboratoriach. RNA-seq pozwala określić ilościowo, odkryć i profilować RNA. Dla tej techniki mRNA (i inne RNA) są najpierw konwertowane do cDNA. CDNA jest następnie używany jako wejście do przygotowania biblioteki sekwencyjnej nowej generacji. W tym artykule przedstawię krótki przegląd RNA-seq i przedstawię główne metody stosowane obecnie.
dlaczego RNA-Seq jest „lepszy” niż Microarrays?,
istnieje kilka zalet RNA-seq nad mikromacierzami:
z RNA-seq można przesłuchać więcej niż tylko różnicową ekspresję genów. Chociaż istnieją mikroarray dostępne do analizy exon-level i microRNA, większość użytkowników nadal jest zainteresowana podstawową, prawdopodobnie 3 ' tendencyjną, różniczkową ekspresją genów. Dzięki RNA-seq można spojrzeć na kodujące i niekodujące RNA, na splicing i ekspresję specyficzną dla alleli, a być może wkrótce na sekwencje cDNA o Pełnej długości, eliminując potrzebę wnioskowania lub składania izoform.,
Microarrays są również stronnicze, ponieważ musimy zdecydować, jaką zawartość umieścić na tablicy. Ponieważ RNA-seq nie używa sond ani podkładów, dane cierpią z powodu znacznie niższych uprzedzeń (chociaż nie chcę powiedzieć, że RNA-seq nie ma żadnych).
RNA-seq dostarcza dane cyfrowe w postaci wyrównanych odczytów, co daje bardzo szeroki zakres dynamiczny, poprawiając czułość wykrywania rzadkich transkryptów.
jest również bardzo konkurencyjny pod względem kosztów w stosunku do mikroarrayów, ponieważ obecnie między 6-30 próbek można multipleksować w jednym torze sekwencyjnym Illumina.,
na koniec możesz ponownie przeanalizować zbiór danych RNA-seq, gdy pojawi się więcej informacji na temat transkryptomu. Jeśli zostanie opublikowany artykuł pokazujący interesujący wariant splicingu w systemie podobnym do tego, nad którym pracujesz, możesz wrócić i spojrzeć na to splicing w swoich próbkach; i masz już dane, aby to zrobić.
jak działa RNA-Seq?
istnieje wiele metod przeprowadzania eksperymentu RNA-seq. W rzeczywistości techniki ewoluują tak szybko, że może być trudno zdecydować, który z nich użyć., Podstawowy wybór to między 1) random-primed synteza cDNA z dwuniciowych cDNA lub 2) metody ligacji RNA (recenzowane i porównywane w Levin 2010). Większość ludzi używa pierwszej metody, a następnie musi dokonać dalszego wyboru między protokołem specyficznym dla danego pasma a protokołem, który nie jest. Metoda stosowana najczęściej w moim laboratorium to TruSeq RNA-seq Illumina, czyli random-primed cDNA synthesis non-strand-specific protocol.
gdy masz bibliotekę sekwencjonowania, jest ona sekwencjonowana do określonej głębokości, która zależy od tego, co chcesz zrobić z danymi., Odczyty te są wyrównane do genomu lub transkryptomu i są liczone w celu określenia ekspresji genu różnicowego lub dalej analizowane w celu określenia splicingu i ekspresji izoform. Większość ludzi sekwencjonuje RNA za pomocą metod 50-100bp. Wyjątkiem jest sekwencjonowanie microRNA, ponieważ wymaga to tylko jednokrotnego sekwencjonowania 36bp w większości przypadków.
nasza metoda RNA-Seq
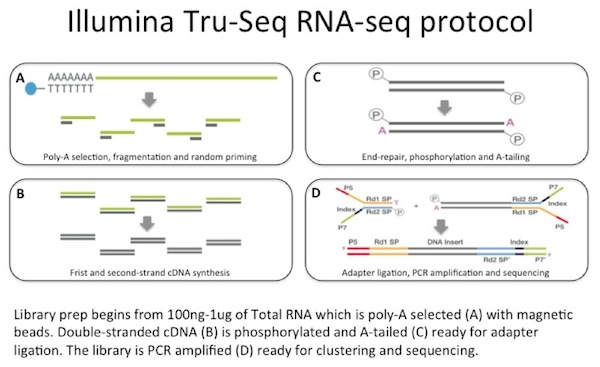
używamy od 100 ng do 1 µg całkowitego RNA jako wejścia do przechwytywania mRNA za pomocą kulek magnetycznych pokrytych oligo-DT. MRNA ulega rozdrobnieniu, a następnie przeprowadza się syntezę cDNA w sposób losowy., Wynikowy dwupasmowy cDNA jest używany jako wejście do standardowego przygotowania biblioteki Illumina, który obejmuje naprawę końcową, ligację adaptera i wzmocnienie PCR, aby zapewnić bibliotekę gotową do sekwencjonowania.
po co zawracać sobie głowę informacją?
było wiele dyskusji na temat transkrypcji anty-sense i jej biologicznego znaczenia. Jeśli jesteś zainteresowany w prostej ekspresji genu różnicowego, a następnie strand informacji nie doda wiele do eksperymentu, ale sprawi, że protokół bardziej skomplikowane., Mimo to, można wykonać najbardziej powszechnie przyjętą metodę bez zbytniego wysiłku. Aby to zrobić, podczas syntezy cDNA drugiej nici, należy użyć uracylu do włączenia zamiast tyminy. Postępuj zgodnie z przygotowaniem Biblioteki Illumina jak zwykle, ale po podwiązaniu adaptera i przed amplifikacją PCR dodaj glikozylazę URACYLO-DNA, aby zniszczyć 2. Strand. Powoduje to, że wszystkie odczyty rozpoczynają się w tej samej orientacji, dzięki czemu można określić, które pasmo było transkrybowane w próbce.
co tak naprawdę można zrobić z RNA-Seq?,
RNA-seq to potężne i wszechstronne narzędzie opublikowane szeroko w ciągu ostatnich kilku lat. Wybrałem kilka moich ulubionych (niektóre z pracy wykonywanej w ośrodku bazowym, którym zarządzam), aby zilustrować, co można zrobić z sekwencjonowaniem RNA.
- Jabbari i in. używane RNA-seq do zbadania łuszczycy i znaleźć nowe geny do analizy funkcjonalnej. Porównali swoje dane RNA-seq do opublikowanych badań tablicy i znaleźli 1700 nowych kandydatów. Zostały one potwierdzone przez qPCR, a porównanie z funkcjonalnymi bazami danych dla łuszczycy potwierdziło ich rolę w patogenezie.
- , wykorzystano RNA-seq w badaniu dotyczącym zachowania wiązania polimerazy RNA III u ssaków w celu walidacji ekspresji genów zajętych przez Pol III w badaniu ChIP-seq.
- połączone wychwytywanie oparte na RNA-seq i microarray w celu identyfikacji i scharakteryzowania rzadkich transkryptów, które są normalnie niewykrywalne. Docelowa liczba transkryptów zwiększyła się z 0,21% przed przechwyceniem do 80% po przechwyceniu., Odkryli ponad 200 wcześniej niezapowiedzianych izoform dla prawie 50 kodujących białka loci, w tym nową alternatywną izoform TP53, która jest bardzo dobrze scharakteryzowanym genem. Sugeruje to, że nadal istnieje wiele złożoności w genomie i transkryptomie do rozwiązania.
podsumowując, RNA-seq jest wciąż rozwijającym się narzędziem, ale w większości przypadków jest preferowany od mikroarrayów. Jest bardziej wrażliwy, bardziej wytrzymały i może być bardziej opłacalny. Jakie projekty RNA-seq planujesz teraz dla swojego projektu?,
Jabbari et al: transkrypcyjne Profilowanie łuszczycy za pomocą RNA-seq ujawnia wcześniej niezidentyfikowane geny o różnej ekspresji. Journal of Investigative Dermatology 2011.
Kutter i wsp.: Wiązanie Pol III u sześciu ssaków wykazuje zachowanie wśród izotypów aminokwasów pomimo różnic w genach tRNA. Nature Genetics 2011
Levin et al: Comprehensive comparative analysis of strand-specific RNA sequencing methods. Metody Przyrodnicze 2010.
Mercer et al: celowane sekwencjonowanie RNA ujawnia głęboką złożoność ludzkiego transkryptomu. Biotechnologia Przyrody 2012.,
Wang et al: RNA-Seq: rewolucyjne narzędzie do transkryptomiki. Nat Rev Genet 2009
Zaktualizowano i poprawiono 16 sierpnia 2015 r.

czy to ci pomogło? Następnie podziel się z siecią.