A totalidade do gene 16S oferece melhor resolução taxonômica
O ~1500 bp 16S rRNA gene é composto por nove variável regiões intercaladas em todo o altamente conservadas 16S sequência (Fig. 1a). Sequenciando todo o gene foi originalmente realizado pela sequenciação de Sanger., Isso exigiu genes de clonagem, gerando, e reunindo duas a três leituras por clone, e produzindo profundidade de amostragem limitada a alto custo e esforço. Atualmente, no entanto, a grande maioria dos estudos seqüência apenas parte do gene, porque a plataforma de sequenciamento ilumina amplamente utilizado (maior rendimento, menor custo, menor esforço em comparação com Sanger) produz sequências curtas ( ≤ 300 bases)., Diferentes sub-regiões do gene são, portanto, orientadas, que vão desde simples variável regiões, tais como V4 ou V6, três variável regiões, tais como V1–V3 ou V3–V5 (usado no Projeto Microbiano Humano em conjunto com a 454 seqüenciamento platform9).
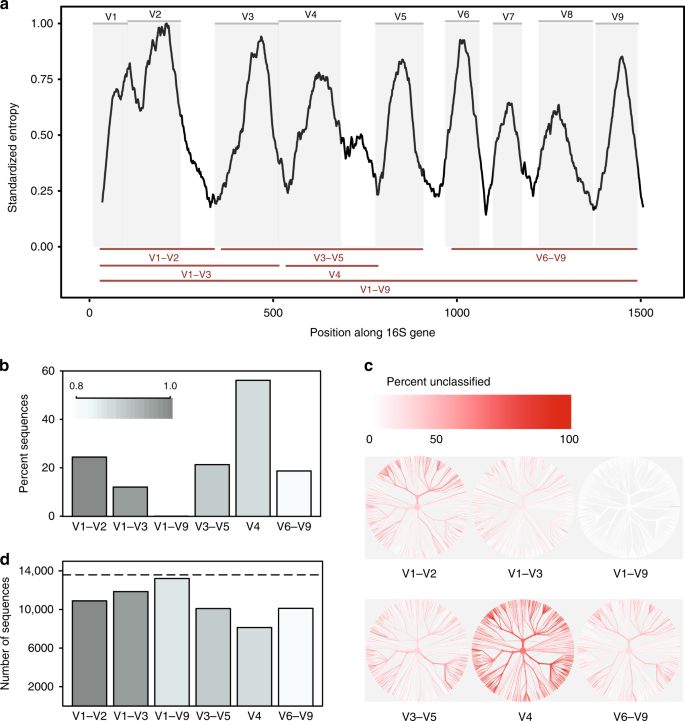
in-silico comparison of 16S rRNA variable regions. uma entropia de Shannon através do gene 16S baseada no alinhamento de uma única sequência representativa para cada espécie conhecida presente no banco de dados Greengenes., As sequências foram alinhadas com um único gene de referência 16S para Escherichia coli K-12 MG1655 (ID do Gene NCBI 947777). Os painéis cinzentos representam as regiões variáveis definidas por locais de ligação de iniciadores geralmente usados (tabela suplementar 1). As regiões variáveis consideradas neste estudo são mostradas como linhas vermelhas (fundo). B proporção de sequências para cada região variável que não pôde ser identificada ao nível da espécie ao classificar cada sequência com base na base de dados de referência da qual foi derivada a um limiar de confiança de 80% (classificador RDP)., C árvores baseadas na taxonomia das sequências presentes na base de dados in-silico. A mesma árvore é fornecida para cada região variável. A cor de cada ramo reflete a proporção de sequências dentro de cada clado que não puderam ser identificadas ao nível da espécie. d O número de OTUs criado ao agrupar sequências para cada região variável com semelhança de sequência de 99%. A linha tracejada indica o número de sequências únicas (>1% diferente) na base de dados original., Dados de origem são fornecidos como uma Fonte de Dados de arquivo
Nós argumentamos que a segmentação em sub-regiões representa um compromisso histórico, devido à tecnologia restrictions10. Hoje, tanto pacbio quanto Oxford Nanopore plataformas de sequenciamento são capazes de rotineiramente produzir leituras em excesso de 1500 bp e sequenciamento de alta capacidade do gene 16S completo está se tornando cada vez mais prevalente., Sugerimos, portanto, que a justificação para este compromisso tenha de ser revista e realizámos uma simples experiência in-silica para demonstrar a vantagem de uma sequenciação 16S completa sobre a orientação das sub-regiões.
baixámos um conjunto de sequências 16S de comprimento completo de uma base de dados pública (Greengenes)., Aproveitando o fato de que uma parte substancial destas sequências constituídas de PCR primer-sítios de ligação, nós aparadas-los para gerar in-silico amplicons para diferentes sub-regiões, com base na localização dos primers de PCR comumente usado no microbiano estudos (Fig. 1a E Quadros suplementares 1-2)., Supondo que cada sequência em nosso download de banco de dados representado uma única espécie, em seguida, usamos uma classificação comum de abordagem (do Ribossoma, o Projeto de Banco de dados (RDP) classifier11) para calcular a frequência com que in-silico amplicons para cada sub-região poderia fornecer informações precisas, espécies de nível de classificação taxonômica (usando o banco de dados original como referência). Numa segunda experiência, também reunimos os nossos amplificadores in-silico para gerar OTUs em diferentes limiares de similaridade de sequências, comummente utilizados.(97%, 98%, 99%).,
descobrimos que sub-regiões diferiam substancialmente na medida em que elas poderiam com confiança discriminar entre as sequências de 16S de comprimento completo usadas para representar espécies (Fig. 1b). A região V4 teve pior desempenho, com 56% De amplicons in-silico falhando em corresponder confiantemente a sua sequência de origem a este nível taxonómico. Em contraste, quando uma sequência de comprimento completo Com todas as regiões variáveis foi usada, foi possível classificar quase todas as sequências como a espécie correta (Figo suplementar. 1a)., Alterar bases de dados e limiares de confiança na classificação afetou a proporção de amplicons in-silico que poderia ser exatamente igualada, mas não influenciou as tendências prevalecentes (Fig suplementar. 1a, b).segundo, diferentes sub-regiões mostraram viés nos taxa bacterianos que foram capazes de identificar (Fig. 1c). Por exemplo, a região V1–V2 teve um desempenho fraco na classificação de sequências pertencentes à Proteobacteria do filo, enquanto a região V3–V5 teve um desempenho fraco na classificação de sequências pertencentes ao filo Actinobacteria (Figo suplementar. 2)., Observaram-se tendências semelhantes a nível do género para os taxa de potencial relevância médica. Apesar de todas as V1–V9 região consistentemente produzido os melhores resultados, o V6–V9 região foi, nomeadamente, o melhor sub-região de classificação de sequências pertencentes aos gêneros Clostridium e Staphylococcus, o V3–V5 região produziu bons resultados para a Klebsiella e a V1–V3 região produziu bons resultados para a Escherichia/Shigella (Complementar Fig. 2 e dados de origem).
finalmente, a escolha da sub-região afetou dramaticamente o número de OTUs formados ao agrupar amplicons in-silico para criar OTUs., Ao agrupar-se a 99% de identidade sequencial, todas as sub-regiões não conseguiram recriar o número de sequências distintas presentes na base de dados original; no entanto, a região V4 novamente realizou o pior (Fig. 1d). Nomeadamente, o número relativo de OT produzidos por cada sub-região não era coerente com diferentes limiares de identidade.(97%, 98%, 99%, Figo suplementar. 3), indicando que o comportamento de algoritmos de agrupamento pode ser difícil de prever quando a quantidade de informação contida dentro de uma região sequenciada é altamente variável.,em conclusão, visar sub-regiões representa um compromisso histórico suficiente para a identificação dos taxa ao nível do género ou acima. No entanto, a nossa simples experiência in-sílica demonstra que não é válido assumir que um agrupamento cada vez mais fino destas sub-regiões resultará na melhoria da resolução taxonómica necessária para reflectir as espécies. Embora algumas sub-regiões (por exemplo, V1–V3) fornecem uma aproximação razoável da diversidade 16S, a maioria não captura variação suficiente de sequência para discriminar entre taxa intimamente relacionados., Notamos também que os polimorfismos discriminatórios podem ser limitados a regiões variáveis específicas; assim, certas sub-regiões serão mais adequadas para discriminar membros estreitamente relacionados de certos taxa.
16S as variantes de cópia genética reflectem a variação do nível de estirpe
a Agrupamento de sequências de 16S em OTUs tem servido historicamente dois objectivos. Em primeiro lugar, removeu pequenas variantes de sequência artificial devido a erros de amplificação e sequenciação da PCR ao colapsar sequências em grupos. Em segundo lugar, colapsou variantes legítimas de sequência que existem entre táxons bacterianos intimamente relacionados., Embora este último possa nem sempre ser desejável, é lógico que você não pode distinguir entre taxa bacteriana cujas 16S sequências variam a uma taxa que é menor do que o erro encontrado em uma determinada plataforma sequenciadora.
recentemente, os avanços no CCS melhoraram drasticamente as taxas de erro das plataformas de sequenciação de longa leitura. Ao mesmo tempo, Métodos Computacionais tornaram possível distinguir entre variação legítima de sequência artificial., Estes avanços tecnológicos e metodológicos significam que os pesquisadores agora têm o potencial de realizar sequenciamento de alto rendimento que pode detectar com precisão variantes de um único nucleótido em todo o gene 16S.
embora seja tentador assumir que variantes de um único nucleótido podem representar táxons distintos, intimamente relacionados, advertimos contra esta interpretação simplista, devido ao fato de que muitos genomas bacterianos contêm múltiplas cópias polimórficas dos GENE12,13,14., Realizamos a sequenciação PacBio CCS de uma comunidade de 36 Espécies de simulação bacteriana (tabela suplementar 3 e Fig suplementar. 4) para demonstrar (i) que a sequência 16S de muitas bactérias varia entre operões dentro do mesmo genoma e (ii) que o sequenciamento de alto rendimento é suficientemente preciso para resolver essas diferenças intergenômicas.
alinhamos as sequências de 16s de PacBio a uma base de dados de referência contendo uma única sequência de 16S representativa para cada membro da nossa comunidade falsa e usamos as estatísticas de alinhamento para avaliar a precisão desta abordagem sequenciadora., Comparando o número de passes é usado para gerar um CCS com a ocorrência de um único nucleotídeo substituições, inserções e exclusões indicou que dez passes poderiam minimizar estes erros combinados a uma frequência mínima de < 1.0% (embora fosse notório que o mínimo realizáveis erro variou entre seqüenciamento é executado; Complementar Fig. 5). No entanto, observamos uma coincidência de erros de eliminação com o local homopolímero executado em nossas sequências de referência (Figo suplementar., 6), que não era específico do nucleótido e foi exacerbado pelo comprimento do homopolímero sequenciado (Figo suplementar. 7). Posteriormente, validámos as deleções no gene Escherichia coli 16S, utilizando a sequência “Lightina whole genome shotgun” (WGS), o que demonstrou que apenas uma das deleções que ocorreram nas sequências de PacBio era genuína (Fig suplementar. 8).,
convencido de que a sequenciação CCS pode produzir leituras 16S com uma baixa frequência de erros de substituição, em seguida, argumentamos que uma proporção dos erros de substituição dentro de leituras alinhadas com precisão deve refletir variação atribuível a 16 polimorfismos dentro de um genome12 espécie. Por exemplo, leituras alinhadas com a estirpe E. coli K-12 substr. MG1655 mostrou um perfil de substituição, que espelhou exatamente o previsto ao alinhar todas as sete sequências de 16S conhecidas por estarem presentes neste genome15(Fig. 2a, c)., Fomos ainda mais capazes de validar a estequiometria destas substituições nucleotídicas quantificando a variação em leituras comparavelmente alinhadas de Illumina WGS (Fig. 2) e demonstrar que um perfil de substituição semelhante era reprodutível em várias séries (Fig. 9)., Alinhamentos para outras sequências de referência em nossa simulação comunidade mostrou-se uma tendência semelhante de abundante substituições localizada específicos da base de dados de posições ao longo do gene 16S, embora nota-se que o sinal-para-ruído aumentou significativamente quando o gene 16S em questão tinha menos de 100 alinhado lê (Complementar Fig. 10).

polimorfismos em sequências genéticas de E. coli 16S rRNA. a a posição e frequência das substituições que aparecem em E., coli estirpe K-12 MG1655 amplicons V1-V9 gerados a partir da nossa comunidade falsa e sequenciados na plataforma PacBio RS II. b a posição e frequência das substituições nas leituras geradas pela sequenciação genómica da estirpe isolada de E. coli K-12 MG1655 na plataforma Illumina MiSeq. As regiões ampliadas mostram as respectivas posições no alinhamento dos sete genes 16S presentes no genoma de referência de E. coli K-12 MG1655. A sequência 16S do rrnd operon ( * * ) é usada como referência para toda a fase SNP. c Perfil de substituição de nucleótidos previsto de E., coli K-12 MG1655 com base no alinhamento das sete sequências genéticas de 16S presentes no genoma de referência. d o perfil de substituição previsto de E. coli O157 Sakai baseado no alinhamento das sete sequências genéticas de 16S presentes no genoma de referência. Os painéis cinzentos representam as regiões variáveis definidas por locais de ligação de iniciadores geralmente usados (tabela suplementar 1). Linhas tracejadas indicam a proporção esperada de substituições nucleotídicas, dado que existem sete cópias de genes 16S dentro de cada genoma., Os dados de origem são fornecidos como um ficheiro de dados de origem
a observação de que a sequenciação de leitura longa pode identificar os polimorfismos de 16S no mesmo genoma tem implicações importantes. Em primeiro lugar,demonstra que não é válido assumir que a sequência de alto rendimento lê diferentes por um ou poucos nucleótidos representam um táxo distinto 6, 16. Dentro de um único genoma, duas ou mais sequências de 16S podem ser idênticas, enquanto outras podem ser únicas., Correspondentemente, alguns loci homólogos de 16S podem manter uma sequência idêntica entre duas estirpes estreitamente relacionadas, enquanto outros podem ter divergido em uma ou poucas posições nucleotídicas. Neste contexto, qualquer comunidade ou de nível taxonômico interpretação de 16S dados devem, idealmente, para o fato de que a abundância relativa de seqüências de 16S decorrentes intimamente relacionados taxa reflete uma combinação linear de (i) a frequência com que cada sequência única é representada através de genomas e (ii) a abundância relativa dos genomas para cada táxon.,
Em segundo lugar, embora a variação da sequência 16S intragenómica complique a análise a nível comunitário, também tem o potencial de aumentar o poder do gene 16S para discriminar entre taxa estreitamente relacionados, porque permite que a comparação baseada em sequências se estenda através de múltiplos loci divergentes. Por exemplo, existe uma variação suficiente de nucleótidos para distinguir a estirpe K-12 MG1655 de E. coli da estirpe enterohemorrágica O157 Sakai (Fig. 2c, d)., Assim, argumentamos que, quando devidamente contabilizadas, múltiplas cópias polimórficas de 16S não são um inconveniente a ser negligenciado, ao invés disso, elas irão permitir que o gene 16S seja usado na análise microbioma de nível de estirpe. Nós também notamos que o poder da variação da seqüência de 16S intragenômicos para discriminar taxa intimamente relacionado é provável que diminua quando sequências parciais de 16S são usadas. Por exemplo, SNPs distinguindo as estirpes de E. coli K-12 MG1655 (Fig. 2c) de O157 Sakai (Fig. 2d) são encontrados nas regiões variáveis V1, V2, V6 e V9.,
16S polimorfismos podem ser resolvidos in vivo
as comunidades microbiológicas são muitas vezes complexas, existentes em diversos ambientes bioquímicos (por exemplo, fezes, saliva, sputum, etc.) e contendo muitas centenas de táxons únicos cuja abundância relativa se estende por uma ampla gama dinâmica. Esta complexidade não está bem representada em experimentos de Comunidade in-silico ou simulado. Nós, portanto, realizamos um experimento adicional para demonstrar que a sequenciação do gene 16S completo, enquanto contabilizando o SNPs intragenômico 16S pode resolver taxa bacteriana intimamente relacionada in vivo.,realizámos a sequenciação PacBio CCS da região V1-V9 para quatro amostras de fezes humanas colhidas em voluntários adultos saudáveis. Para comparação, sequenciamos a região V1–V3 usando o Illumina MiSeq e, para fornecer uma referência para quantificação taxonômica de nível de espécie, realizamos sequenciação metagenômica WGS (mWGS) usando o Illumina NextSeq. Para avaliar a medida em que cada uma dessas abordagens sequenciadoras pode resolver táxons intimamente relacionados, focamos no gênero Bacteroides., Além de ser abundante no intestino humano, este gênero é altamente diversificado, contendo várias espécies que podem exercer efeitos bons e ruins na saúde humana 17. Ele também tem sido usado anteriormente como um táxon modelo para demonstrar a utilidade do gene 16S para análise taxonômica de alta resolução 18.quando calculámos a abundância de Bacteroides ao nível do género, a sequenciação V1–V9 e a sequenciação V1–V3 produziram resultados comparáveis., Ambas as abordagens identificaram dois indivíduos com baixa abundância relativa de Bacteroides (~10-25%) e dois indivíduos com elevada abundância relativa de Bacteroides (~40-60%; Fig. 3a). No entanto, a quantificação a nível das espécies através da sequenciação mWGS revelou uma diversidade muito maior, com diferentes espécies de Bacteroides dominantes no intestino de cada indivíduo (Fig. 3b e dados suplementares 1). Ao agrupar OTUs a 99% de identidade, tanto a sequenciação V1–V9 como a sequenciação V1–V3 foram capazes de refletir esta variação ao nível da espécie (Fig., 3b), com a notável exceção de que a sequenciação V1–V3 não detectou Bacteroides intestinalis, que era abundante em uma das quatro amostras de microbiomas intestinais humanos. Com base nestes resultados, concluímos que, quando utilizados em conjunto com um limiar de identidade adequado (por exemplo, 99%), abordagens baseadas em OTU têm o potencial de resolver a diversidade de espécies observada no intestino humano. Notamos ainda que, embora a sequenciação 16S de comprimento completo possa ser ideal para a análise de nível de espécie, regiões variáveis altamente informativas (por exemplo, V1-V3) também podem ser adequadas para este propósito.,

detectando Bacteroides em amostras de fezes humanas. A A abundância relativa do género Bacteroides em quatro amostras de fezes humanas quantificadas utilizando amplicons V1–V9 (eixo x) ou amplicons V1-V3 (eixo y). b A abundância relativa de espécies de Bacteroides nas mesmas quatro amostras. A abundância de Espécies foi quantificada a partir da sequenciação de mWGS ou a partir do OTUs V1–V3/V1–V9 gerado a 99% de identidade., A abundância é mostrada para as espécies mais abundantes quantificadas por mWGS (para estimativas de abundância de todas as espécies de Bacteroides detectadas por cada plataforma, ver tabela suplementar 5). C perfis de substituição de nucleótidos gerados pelo alinhamento de todas as sequências de amplificação V1–V9 atribuídas à única OTU identificada como Bacteroides vulgatus. Os perfis são mostrados para as duas amostras de fezes com alta abundância relativa de B. vulgatus (IronHorse e Scott). d perfis de substituição de nucleótidos previstos a partir dos genomas de referência de duas estirpes diferentes de B. vulgatus ATCC 848239 e mpk40., Em ambos c E d, as substituições nucleotídicas foram identificadas em relação a um único gene de referência 16S para B. vulgatus ATCC 8482 (NCBI Gene ID 5304800). Os painéis cinzentos representam as regiões variáveis definidas por locais de ligação de iniciadores geralmente usados (tabela suplementar 1). Linhas tracejadas indicam a proporção esperada de substituições nucleotídicas, dado que existem sete cópias de genes 16S dentro de cada genoma., Dados de origem são fornecidos como uma Fonte de Dados de arquivo
aproveitando o fato de que Bacteroides vulgatus estava presente em elevada abundância relativa em dois de nossos intestino humano microbiano amostras, nós próxima perguntado se intragenomic variação entre gene 16S cópias poderiam ser detectados in vivo. Alinhamos todas as sequências de comprimento completo classificadas como pertencentes ao nosso B. vulgatus V1–V9 OTUs (Fig. 3b e dados suplementares 1) a um único representante sequência genética B. vulgatus 16S. Em seguida, comparamos os perfis de substituição nucleotídica resultantes (Fig., 3c) com perfis previstos a partir de dois genomas de referência presentes na base de dados NCBI Refseq19 (Fig. 3d).
a maioria das variações nucleotídicas presentes em nossa geração in vivo B. vulgatus OTU refletiu variação verdadeira atribuível a polimorfismos intragenômicos. Em contraste, a variação provável devido a erros de sequenciação apareceu baixa e bem abaixo da frequência mínima de ~14% que seria esperado se houvesse uma única estirpe B. vulgatus em cada amostra com sete cópias do gene 16S em seu genoma(Fig. 3C, linhas tracejadas).apesar de não sabermos o número verdadeiro de B., estirpes vulgatus presentes em cada amostra in vivo, foi notável que ambos os perfis de substituição de nucleótidos tinham mais semelhança com a estirpe ATCC 8482 do que mpk. A variação também existia em loci específico que poderia potencialmente indicar diferenças significativas entre os genomas de referência in vivo e ATCC 8482. Por exemplo, um único polimorfismo foi detectado na região V5 do ATCC 8482, que estava presente em três cópias 16S (43%). Na primeira amostra in vivo (Scott) este polimorfismo estava presente em 84% das leituras, enquanto na segunda (IronHorse) estava presente em 69% das leituras., Estes números correspondem De Perto aos números esperados se um polimorfismo estivesse presente Seis e cinco de sete genes 16S, respectivamente.
Em conclusão, nós mostramos que a sequenciação 16S de comprimento completo do microbioma intestinal humano pode resolver com precisão substituições de nucleótidos únicos que refletem a variação intra-genómica entre 16 cópias do gene. A presença de tal variação indica que as sequências de 16S devem ser agrupadas para refletir unidades taxonômicas significativas., Usando OTUs agrupados em 99% de identidade, nós mostramos que 16S de comprimento completo tem o potencial de fornecer espécies e até mesmo resolução taxonômica de nível de tensão. A análise das comunidades microbianas nesses níveis taxonômicos promete fornecer uma perspectiva muito diferente da proporcionada pelas estimativas de abundância de gênero.
Intragenomic 16S polimorfismos são altamente prevalentes
de Ter demonstrado que é possível resolver intragenomic cópia variantes in vivo, o próximo procurou estabelecer em que medida tais cópia variantes aparecem na taxa comumente encontrada no intestino humano microbiano., Procurámos ainda determinar se esses perfis podem ser utilizados rotineiramente para distinguir entre estirpes da mesma espécie.cultivámos 381 taxa a partir do microbioma intestinal dos indivíduos saudáveis representados na Fig. 3, bem como de outras pessoas que participam no mesmo estudo20 original (dados suplementares 2). Subsequentemente realizamos sequenciamento de genes 16S em isolados e leituras sequenciadas alinhadas para identificar substituições nucleotídicas características das variantes de cópia genética 16S.,
classificação taxonómica de isolados identificados 58 espécies putativas (dados suplementares 2), enquanto se agrupa uma única sequência representativa para cada isolado em 99% de semelhança resultou em 61 OTUs (com entre 1 e 73 isolados atribuídos a cada OTU). No total, 349 de 381 isolados sequenciados (54 de 61 OTUs) tinham uma ou mais SNP, indicando a presença de polimorfismos de genes 16S, e 205 perfis SNP únicos foram identificados quando contabilizavam possíveis erros de sequenciação (Fig. 4a e dados suplementares 2).

polimorfismos do gene 16s Intravenómicos em isolados de microbiomas intestinais humanos. uma localização de SNPs presentes nos genes 16S de isolados bacterianos cultivados individualmente. As localizações SNP foram identificadas através de sequências de genes de 16S de comprimento total geradas para cada isolado individual. O eixo X representa a posição ao longo do gene 16S. Axis Y denota isolados individuais agrupados com base em sua filogenia inferida. A região azul escura indica a localização de um polimorfismo., Para maior clareza, é apresentado um máximo de cinco isolados pertencentes à mesma espécie. Para detalhes dos perfis de substituição de nucleótidos para todos os isolados sequenciados, ver dados suplementares 2. exemplos b-d de perfis de substituição de nucleótidos que mostram diferenças de nível de estirpe entre isolados identificados como pertencentes a três espécies bacterianas: B Shigella flexneri; C Bifidobacterium longum; d Collinsella aerofaciens. Para cada espécie, dois perfis de substituição de nucleótidos isolados são mostrados; no entanto, exemplos adicionais podem ser encontrados em dados suplementares 2., Os isolados foram identificados como pertencentes à mesma espécie se as suas sequências representativas fossem atribuídas à mesma OTU quando se agrupavam a 99% de identidade sequencial. A identificação taxonômica foi realizada usando BLAST para alinhar sequências representativas ao banco de dados BLAST NCBI 16S (ver Métodos). Os painéis cinzentos representam as regiões variáveis definidas por locais de ligação de iniciadores geralmente usados (tabela suplementar 1). As linhas tracejadas indicam a proporção esperada de substituições nucleotídicas, dado o número de 16 cópias genéticas previstas para cada genoma., Dados de origem são fornecidos como uma Fonte de Dados de arquivo
Nomeadamente, comparando SNP perfis para isola atribuídos à mesma OTU frequentemente revelou diferenças na freqüência de SNPs que foram sugestivos de diferenças em intragenomic gene 16S cópias entre intimamente relacionados táxons. Exemplos de diferentes perfis de substituição são apresentados para três taxa (Fig. 4b-d), sugestivas de variação do nível da estirpe comparável à que demonstrámos em princípio para a E. coli (Fig. 2b).,
Em conclusão, mostramos que muitos dos membros culturáveis do microbioma intestinal humano possuem frequentemente polimorfismos de genes 16S, que, quando devidamente contabilizados, têm o potencial de resolver estirpes da mesma espécie.