RNA (Wang 2009) está rapidamente substituindo microarray de expressão genética em muitos laboratórios. RNA-seq permite quantificar, descobrir e traçar RNAs. Para esta técnica, o ARNm (e outros RNAs) são convertidos pela primeira vez em cDNA. O cDNA é então usado como entrada para uma biblioteca de sequenciamento de próxima geração. Neste artigo, vou fazer uma breve revisão do RNA-seq e introduzir os principais métodos que estão sendo usados hoje.porque é que o RNA-Seq é “melhor” do que os Microarrays?,
Existem várias vantagens que o RNA-seq tem sobre os microarrays:
com RNA-seq você pode interrogar mais do que apenas a expressão diferencial do gene. Embora existam microarrays disponíveis para análise de nível exon e microRNA, a maioria dos usuários ainda estão interessados na expressão genética diferencial básica, provavelmente 3′ tendenciosa. Com RNA-seq você pode olhar para a codificação e não-codificação RNA, na expressão específica de splicing e Alelo, e possivelmente em breve em sequências cDNA de comprimento completo, eliminando a necessidade de inferir ou montar isoformas.,
Microarray também são tendenciosos, uma vez que temos que decidir que Conteúdo colocar no array. Uma vez que RNA-seq não usa sondas ou primers, os dados sofrem de vieses muito menores (embora eu não quero dizer que RNA-seq não tem nenhuma).
RNA-seq fornece dados digitais na forma de contagens de leitura alinhadas, resultando em uma ampla gama dinâmica, melhorando a sensibilidade de detecção para transcrições raras.
também é muito rentável para microarrays, como hoje, entre 6-30 amostras podem ser multiplexadas em uma única faixa de sequenciação ilumina.,por último, pode reanalisar um conjunto de dados RNA-seq à medida que mais informação sobre o transcriptoma se torna disponível. Se um artigo é publicado mostrando uma interessante variante do splice em um sistema semelhante ao que você trabalha, então você pode querer voltar e olhar para esse splicing em suas amostras; e você já teria os dados para fazê-lo.como funciona o RNA-Seq?
Existem muitos métodos para realizar um experimento RNA-seq. Na verdade, as técnicas estão evoluindo tão rapidamente que pode ser difícil decidir qual usar., Uma escolha básica é entre 1) síntese aleatória de cDNA a partir de métodos de dupla cadeia cDNA ou 2) Rna-ligação (revisada e comparada em Levin 2010). A maioria das pessoas utiliza o primeiro método e, em seguida, precisa fazer uma nova escolha entre um protocolo específico da vertente e um que não é. O método mais utilizado no meu laboratório é o ARN-seq de Illumina’s TruSeq, que é um protocolo de síntese de cDNA não-específico de cadeia aleatória.
Uma vez que você tem uma biblioteca de sequenciamento, ela é sequenciada para uma profundidade especificada, que é dependente do que você quer fazer com os dados., Estas leituras estão alinhadas ao genoma ou transcriptoma e são contadas para determinar a expressão diferencial do gene ou ainda analisadas para determinar a expressão splicing e isoforma. A maioria das pessoas está sequenciando RNA usando métodos emparelhados de 50-100bp. A exceção é a sequenciação de microRNA, pois isso só requer sequenciação de 36bp de ponta única na maioria dos casos.
o nosso método RNA-Seq
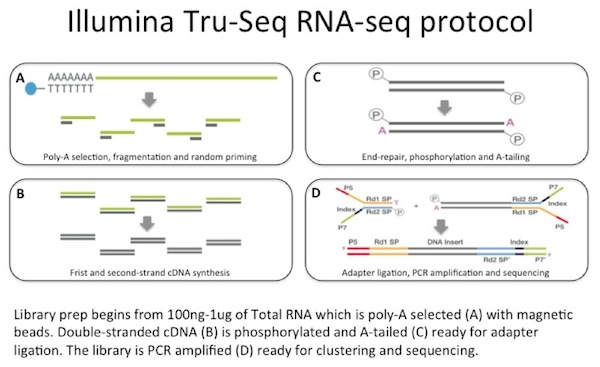
usamos entre 100 ng e 1 µg de RNA total como entrada para uma captura de mRNA com esferas magnéticas revestidas de oligo-dT. O ARNm é fragmentado e, em seguida, uma síntese de cDNA de teste aleatório é realizada., O cDNA duplo resultante é usado como entrada para uma biblioteca padrão Illumina que inclui reparação final, ligação adaptadora e amplificação PCR para lhe dar uma biblioteca pronta para sequenciação.
porquê preocupar-se com a informação da cadeia de caracteres?
tem havido muita discussão sobre a transcrição anti-sensorial e a sua relevância biológica. Se você está interessado na expressão de genes diferenciais simples, então a informação strand não vai adicionar muito à sua experiência, mas vai tornar o seu protocolo mais complexo., Dito isto, você pode executar o método mais amplamente adotado sem muito esforço extra. Para isso, durante a síntese de cDNA da 2ª cadeia, use uracilo para incorporação em vez de timina. Siga o Illumina library prep Como normal, mas após a ligação do adaptador e antes da amplificação PCR adicionar uracil-DNA glicosilase para degradar o 2ndstrand. Isso resulta em todas as leituras começando na mesma orientação para que você possa determinar qual strand estava sendo transcrito em sua amostra.
o que você pode realmente fazer com RNA-Seq?,
RNA-seq é uma ferramenta poderosa e versátil publicada amplamente nos últimos anos. Eu escolhi um par de meus favoritos (alguns do trabalho realizado no núcleo da instalação que eu gerencio) para ilustrar o que você pode fazer com Rna-sequenciamento.
- Jabbari, et al. utilizou RNA-seq para investigar a psoríase e encontrar novos genes para análise funcional. Eles compararam seus dados RNA-seq com os estudos array publicados e encontraram 1700 novos candidatos. Estes foram validados pela qPCR, e a comparação com bases de dados funcionais para psoríase suportou seu papel na patogênese.Kutter, et al., usado RNA-seq em um estudo que analisa a conservação da ligação RNA polimerase III em mamíferos para validar a expressão de genes ocupados por Pol III como assentado por ChIP-seq.Mercer, et al. combined RNA-seq and microarray-based capture to identify and characterize rare transcripts, which are normally undetectable. As transcrições aumentaram em seqüência de abundância de 0,21% pré-captura para 80% pós captura., Eles encontraram mais de 200 isoformas previamente não anotadas para quase 50 loci codificadores de proteínas, incluindo uma nova isoforma alternativa de TP53, que é um gene muito bem caracterizado. Isto sugere que ainda há muita complexidade no genoma e transcriptoma a ser resolvido.
em resumo, RNA-seq ainda é uma ferramenta em evolução, mas é preferível na maioria dos casos aos microarrays. É mais sensível, mais robusta e pode ser mais rentável. Que projectos RNA-seq está a planear para o seu projecto?,Jabbari et al: o perfil Transcritional da psoríase usando RNA-seq revela Genes previamente não identificados e expressos de forma diferente. Journal of Investigative Dermatology 2011.
Kutter et al: a ligação Pol III em seis mamíferos mostra conservação entre os isotipos de aminoácidos, apesar da divergência entre os genes tRNA. Nature Genetics 2011.
Levin et al: Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nature Methods 2010.
Mercer et al: a sequenciação de RNA alvo revela a profunda complexidade do transcriptoma humano. Nature Biotechnology 2012.,Wang et al: RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009.
originalmente publicado em 25 de Maio de 2012. Atualizado e revisto em 16 de agosto de 2015.

isto ajudou-o? Então, por favor, partilhe com a sua rede.