complet De gene 16S oferă o clasificare taxonomică
~1500 bp genei pentru arnr 16S cuprinde nouă variabilă regiuni intercalate pe parcursul înalt conservată secvența 16S (Fig. 1a). Secvențierea întregii gene a fost realizată inițial prin secvențierea Sanger., Aceasta a necesitat clonarea genelor, generarea și asamblarea a două până la trei citiri pe clonă și producerea unei adâncimi de eșantionare limitate la costuri și efort ridicat. În prezent, însă, marea majoritate a studiilor secvențiază doar o parte a genei, deoarece platforma de secvențiere Illumina utilizată pe scară largă (debit mai mare, cost mai mic, efort redus comparativ cu Sanger) produce secvențe scurte ( ≤ 300 de baze)., Diferite sub-regiuni ale genei, prin urmare, sunt vizate, variind de la o singură variabilă regiuni, cum ar fi V4 sau V6, la trei variabile regiuni, cum ar fi V1–V3 sau V3–V5 (utilizate în Proiectul Microbiomului Uman, în colaborare cu 454 de secvențiere platform9).
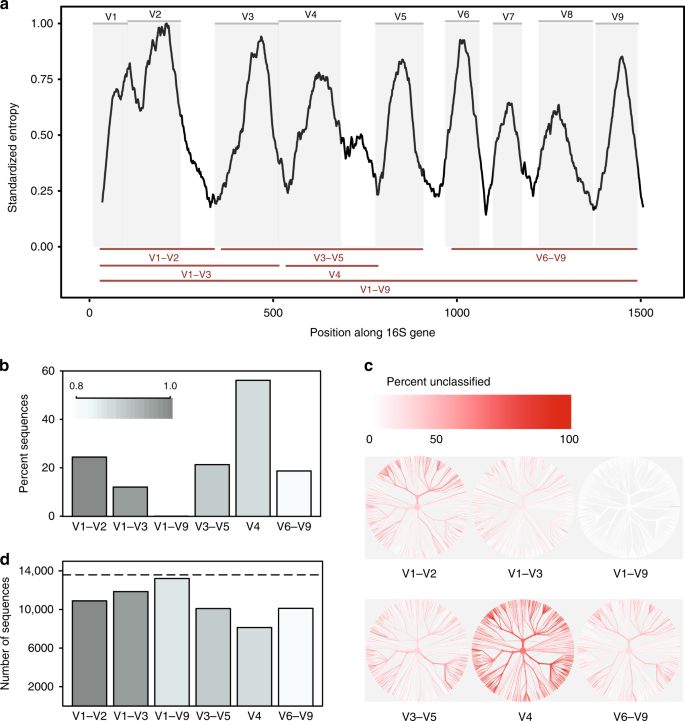
In-silico compararea 16S arnr variabilă regiuni. o entropie Shannon peste gena 16S bazat pe alinierea unei singure secvențe reprezentative pentru fiecare specie cunoscută prezentă în baza de date Greengenes., Secvențele au fost aliniate cu o singură genă de referință 16S pentru Escherichia coli K-12 MG1655 (Gena NCBI ID 947777). Panourile gri prezintă regiuni variabile definite de locurile de legare a grundurilor utilizate în mod obișnuit (tabelul suplimentar 1). Regiunile variabile luate în considerare în acest studiu sunt prezentate ca linii roșii (partea de jos). B proporția secvențelor pentru fiecare regiune variabilă care nu au putut fi identificate la nivel de Specie la clasificarea fiecărei secvențe în baza de date de referință din care a fost derivată la un prag de încredere de 80% (clasificator RDP)., c arbori pe baza taxonomiei secvențelor prezente în baza de date in-silico. Același arbore este prevăzut pentru fiecare regiune variabilă. Culoarea fiecărei ramuri reflectă proporția secvențelor din fiecare cladă care nu au putut fi identificate la nivelul speciilor. d numărul de Otu-uri create atunci când se grupează secvențe pentru fiecare regiune variabilă la similitudinea secvenței de 99%. Linia punctată indică numărul de secvențe unice (>1% diferit) în baza de date originală., Sursa de date sunt furnizate ca o Sursă fișier de Date
Vom argumenta că vizează sub-regiuni reprezintă un compromis istoric, datorită tehnologiei restrictions10. Astăzi, platformele de secvențiere nanopore PacBio și Oxford sunt capabile să producă în mod obișnuit citiri care depășesc 1500 bp, iar secvențierea cu randament ridicat a genei complete 16S devine din ce în ce mai răspândită., Prin urmare, sugerăm că justificarea acestui compromis trebuie revizuită și am efectuat un simplu experiment in-silico pentru a demonstra avantajul secvențierii 16S pe toată lungimea față de direcționarea subregiunilor.
Am descarcat un set de non-redundante (de exemplu, > 1% diferite), full-lungime 16 secvențe dintr-o bază de date publică (Greengenes)., Profitând de faptul că o proporție substanțială din aceste secvențe încorporate PCR primer-site-uri de legare, am tuns-le pentru a genera in-silico ampliconilor pentru diferite sub-regiuni, în funcție de locația de primeri PCR frecvent utilizate în microbiome studii (Fig. 1a și tabelele suplimentare 1-2)., Presupunând că fiecare secvență în noastre de date descărcate reprezentat o specie unică, apoi am folosit o clasificare comună abordare (Ribozomului baza de Date a Proiectului (RDP) classifier11) pentru a calcula frecvența cu care in-silico ampliconilor pentru fiecare sub-regiune ar putea oferi exacte, specii-nivel încadrarea taxonomică (folosind baza de date originală, ca o referință). Într-un al doilea experiment, am grupat, de asemenea, ampliconii noștri in-silico pentru a genera Otu la diferite praguri de similitudine a secvențelor utilizate în mod obișnuit (97%, 98%, 99%).,
am constatat că subregiunile diferă substanțial în măsura în care ar putea discrimina cu încredere între secvențele 16S de lungime întreagă folosite pentru a reprezenta speciile (Fig. 1b). Regiunea V4 s-a comportat cel mai rău, 56% dintre ampliconii in-silico nereușind să se potrivească cu încredere cu secvența lor de origine la acest nivel taxonomic. În schimb, atunci când a fost utilizată o secvență de lungime întreagă cu toate regiunile variabile, a fost posibilă clasificarea aproape a tuturor secvențelor ca specie corectă (Fig suplimentar. 1a)., Modificarea bazelor de date și a pragurilor de încredere în clasificare au afectat proporția ampliconilor in-silico care ar putea fi corect corelați, dar nu au influențat tendințele predominante (Fig. 1A, b).în al doilea rând, diferite subregiuni au prezentat părtinire în taxonii bacterieni pe care au putut să-i identifice (Fig. 1C). De exemplu, V1–V2 regiune rezultate slabe la clasificarea secvențe aparținând încrengătura Proteobacteria, întrucât V3–V5 regiune rezultate slabe la clasificarea secvențe aparținând încrengătura Actinobacteria (Suplimentare Fig. 2)., Tendințe similare au fost observate la nivelul genului pentru taxoni cu relevanță medicală potențială. Deși full V1–V9 regiune produs în mod constant cele mai bune rezultate, V6–V9 regiune a fost ales cel mai bun sub-regiune pentru clasificarea secvențe aparținând genurilor Clostridium, Staphylococcus, V3–V5 regiune a produs rezultate bune pentru Klebsiella, și V1–V3 regiune a produs rezultate bune pentru Escherichia/Shigella (Suplimentare Fig. 2 și date sursă).în cele din urmă, alegerea subregiunii a afectat dramatic numărul de Otu-uri formate la gruparea ampliconilor in-silico pentru a crea Otu-uri., Când se grupează la identitatea secvenței de 99%, toate subregiunile nu au reușit să recreeze numărul de secvențe distincte prezente în baza de date originală; cu toate acestea, regiunea V4 a avut din nou cel mai rău rezultat (Fig. 1D). În special, numărul relativ de Otu produse de fiecare subregiune nu a fost consecvent la praguri de identitate diferite(97%, 98%, 99%, Fig suplimentar. 3), indicând faptul că comportamentul algoritmilor de grupare poate fi dificil de prezis când cantitatea de informații conținute într-o regiune secvențiată este foarte variabilă.,în concluzie, vizarea subregiunilor reprezintă un compromis istoric care a fost suficient pentru identificarea taxonilor la nivelul genului sau mai sus. Cu toate acestea, experimentul nostru simplu in-silico demonstrează că nu este valabil să presupunem că gruparea tot mai fină a acestor subregiuni va duce la o rezoluție taxonomică îmbunătățită necesară pentru a reflecta speciile. Deși unele subregiuni (de exemplu, V1-V3) oferă o aproximare rezonabilă a diversității 16S, majoritatea nu captează suficiente variații de secvență pentru a discrimina taxonii strâns legați., De asemenea, observăm că polimorfismele discriminatorii pot fi limitate la anumite regiuni variabile; astfel, anumite subregiuni vor fi mai potrivite pentru discriminarea membrilor strâns legați ai anumitor taxoni.
variantele de copiere a genelor 16S reflectă variația nivelului tulpinii
gruparea secvențelor 16S în OTUs a servit istoric în două scopuri. În primul rând, a eliminat variante minore de secvență artifactuală din cauza amplificării PCR și a erorilor de secvențiere la prăbușirea secvențelor în grupuri. În al doilea rând, s-a prăbușit variante de secvență legitime care există între taxoni bacterieni strâns legați., Deși acesta din urmă nu poate fi întotdeauna de dorit, este evident că nu puteți distinge între taxonii bacterieni ale căror secvențe 16S variază cu o rată mai mică decât eroarea întâlnită pe o anumită platformă de secvențiere.recent, progresele înregistrate în CSC au îmbunătățit dramatic ratele de eroare ale platformelor de secvențiere cu citire lungă. În același timp, metodele computaționale au făcut posibilă distingerea între variația legitimă vs.artifactuală a secvenței., Aceste progrese tehnologice și metodologice înseamnă că cercetătorii au acum potențialul de a efectua secvențiere de mare viteză care poate detecta cu precizie variantele cu un singur nucleotid pe întreaga genă 16S.
Deși este tentant să presupunem că single-nucleotide variante pot reprezenta distincte, strâns legate de taxoni, ne precauție împotriva acestei mult prea simplistă interpretare datorită faptului că multe genomuri bacteriene conțin mai multe polimorfă copii de 16 gene12,13,14., Am efectuat secvențierea PacBio CCS a unei comunități mock bacteriene de 36 de specii (tabelul suplimentar 3 și Fig suplimentar. 4) să demonstreze: (i) că 16S secvență de multe bacterii variază între operons în același genom și (ii) că high-throughput secvențiere este suficient de precisă pentru a rezolva aceste intragenomic diferențe.
am aliniat secvențele PacBio full-length 16S la o bază de date de referință care conține o singură secvență reprezentativă 16S pentru fiecare membru al comunității noastre mock și am folosit statisticile de aliniere pentru a evalua acuratețea acestei abordări de secvențiere., Comparând numărul de treceri utilizat pentru a genera o CCS cu apariția single-nucleotide substituții, inserări și ștergeri a indicat că zece treceri putea minimiza aceste combinate erori la o frecvență minimă de < 1.0% (deși a fost notabile minim realizabil eroare a variat între secvențiere ruleaza; Suplimentar Fig. 5). Cu toate acestea, am observat o coincidență a erorilor de ștergere cu homopolimerul locației care rulează în secvențele noastre de referință (Fig suplimentar., 6), care nu a fost specifică nucleotidelor și a fost exacerbată de lungimea homopolimerului secvențiat (Fig suplimentar. 7). Ulterior, am validat ștergerile din gena Escherichia coli 16S folosind secvențierea Illumina whole genome shotgun( WGS), care a demonstrat că doar una dintre ștergerile care apar în secvențele PacBio a fost autentică (Fig. 8).,
Mulțumit de faptul că CSC secvențiere poate produce 16 citește cu o frecvență scăzută de erori de substituire, avem următoarea motivat că un procent de erori de substituire în cadrul aliniate cu precizie citeste ar trebui să reflecte variația atribuite 16 polimorfisme în cadrul unei specii’ genome12. De exemplu, citește aliniat la tulpina E. coli K-12 substr. MG1655 a arătat un profil de substituție, care a reflectat exact ceea ce a prezis prin alinierea tuturor celor șapte secvențe 16S cunoscute a fi prezente în acest genom15 (Fig. 2a, c)., Am reușit în continuare să validăm stoichiometria acestor substituții de nucleotide prin cuantificarea variației în citirile Illumina WGS aliniate comparabil (Fig. 2B) și să demonstreze că un profil de substituție similar a fost reproductibil pe mai multe serii de secvențiere (Fig. 9)., Aliniamentele la alte secvențe de referință din comunitatea noastră mock au arătat o tendință similară de substituții abundente localizate în poziții de bază specifice de-a lungul genei 16S, deși observăm că raportul semnal-zgomot a crescut semnificativ atunci când gena 16S în cauză a avut mai puțin de 100 de citiri aliniate (Fig suplimentar. 10).

Polimorfisme în E. coli genei pentru arnr 16S. a poziția și frecvența substituțiilor care apar în E., coli tulpina K-12 mg1655 V1–V9 amplicons generate de comunitatea noastră mock și secvențiate pe platforma PacBio RS II. b poziția și frecvența de substituții în citește generate de secvențiere genomică de izolată tulpina de E. coli K-12 MG1655 pe Illuminati MiSeq platforma. Regiunile mărite prezintă pozițiile respective în alinierea tuturor celor șapte gene 16S prezente în genomul de referință E. coli K-12 MG1655. Secvența 16S din operonul rrnD (**) este utilizată ca referință pentru toate fazele SNP. c profilul anticipat de substituție nucleotidică al E., coli K-12 MG1655 pe baza alinierii celor șapte secvențe genice 16S prezente în genomul de referință. d profilul de substituție prezis al E. coli O157 Sakai bazat pe alinierea celor șapte secvențe genice 16S prezente în genomul de referință. Panourile gri prezintă regiuni variabile definite de locurile de legare a grundurilor utilizate în mod obișnuit (tabelul suplimentar 1). Liniile punctate indică proporția așteptată de substituții de nucleotide, având în vedere că există șapte copii ale genei 16S în fiecare genom., Datele sursă sunt furnizate ca fișier de date sursă
observația că secvențierea cu citire lungă poate identifica polimorfismele 16S în cadrul aceluiași genom are implicații importante. În primul rând, demonstrează că nu este valabil să se presupună că secvența de mare debit citește diferite de una sau câteva nucleotide reprezintă un taxa6 distinct,16. Într-un singur genom, două sau mai multe secvențe 16S pot fi identice, în timp ce altele pot fi unice., În mod corespunzător, unii loci 16S omologi pot păstra secvențe identice între două tulpini strâns legate, în timp ce alții pot fi Divergenți la una sau câteva poziții nucleotidice. În acest context, orice nivel comunitar sau taxonomice interpretare de 16 date în mod ideal, ar trebui să se țină seama de faptul că abundența relativă de 16 secvențe care decurg din foarte strâns legate de taxoni va reflecta o combinație liniară de (i) frecvența cu care fiecare secvență unică este reprezentată pe genomuri și (ii) abundența relativă de genomuri pentru fiecare taxon.,
în al Doilea rând, deși intragenomic 16 secvență de variație complică la nivel comunitar, analiza, de asemenea, are potențialul de a crește puterea de gene 16S să discrimineze între strâns legate de taxoni, deoarece permite secvență pe baza de comparație să se extindă pe mai multe divergente loci. De exemplu, suficient de nucleotide există variații de a distinge tulpina de E. coli K-12 MG1655 din enterohemorrhagic tulpina O157 Sakai (Fig. 2C, d)., Astfel, argumentăm că, atunci când sunt contabilizate în mod corespunzător, copiile multiple polimorfe 16S nu reprezintă un inconvenient care trebuie trecut cu vederea, ci vor permite ca gena 16S să fie utilizată în analiza microbiomului la nivel de tulpină. De asemenea, observăm că puterea variației secvenței intragenomice 16S de a discrimina taxonii strâns legați este probabil să se diminueze atunci când se utilizează secvențe parțiale 16S. De exemplu, SNP-urile disting tulpinile de E. coli K-12 MG1655 (Fig. 2c) din O157 Sakai (Fig. 2D) se găsesc în regiunile variabile V1, V2, V6 și V9.,
polimorfismele 16S pot fi rezolvate in vivo
comunitățile Microbiome sunt adesea complexe, existente în diverse medii biochimice (de exemplu, scaun, salivă, spută etc.) și care conține multe sute de taxoni unici a căror abundență relativă se întinde pe o gamă dinamică largă. Această complexitate nu este bine reprezentată nici în silico, nici în experimentele comunitare simulate. Prin urmare, am efectuat un experiment suplimentar pentru a demonstra că secvențierea completă 16 gene în timp ce contabile pentru intragenomic 16 Snp poate rezolva strâns legate bacteriene taxoni in vivo.,am efectuat secvențierea PacBio CCS a regiunii V1-V9 pentru patru probe de scaun uman colectate de la voluntari adulți sănătoși. Pentru comparație, am secvențat V1–V3 regiune folosind Illumina MiSeq și, pentru a oferi un punct de referință pentru specii-nivel taxonomic cuantificare, am efectuat metagenomic WGS (mWGS) secvențierea folosind Illumina NextSeq. Pentru a evalua măsura în care fiecare dintre aceste abordări de secvențiere poate rezolva taxoni strâns legați, ne-am concentrat pe genul Bacteroides., Pe lângă faptul că este abundent în intestinul uman, acest gen este foarte divers, conținând mai multe specii care pot exercita atât efecte bune, cât și rele asupra sănătății umane17. De asemenea, a fost utilizat anterior ca taxon model pentru demonstrarea utilității genei 16S pentru analiza taxonomică de înaltă rezoluție18.când am calculat abundența Bacteroidelor la nivelul genului, secvențierea V1–V9 și secvențierea V1–V3 au produs rezultate comparabile., Ambele abordări au identificat doi indivizi cu abundență relativă scăzută a Bacteroidelor (~10-25%) și doi indivizi cu abundență relativă ridicată a Bacteroidelor (~40-60%; Fig. 3a). Cu toate acestea, cuantificarea la nivel de specie prin secvențiere mWGS a evidențiat o diversitate mult mai mare, cu o specie Bacteroides diferită dominantă în intestinul fiecărui individ (Fig. 3b și date suplimentare 1). Când se grupează OTUs la identitate de 99%, atât secvențierea V1–V9, cât și secvențierea V1–V3 au putut reflecta această variație a nivelului speciilor (Fig., 3B), cu excepția notabilă că secvențierea V1–V3 nu a detectat Bacteroides intestinalis, care a fost abundent într-una din cele patru probe de microbiom intestinal uman. Pe baza acestor rezultate, concluzionăm că, atunci când sunt utilizate împreună cu un prag de identitate adecvat (de exemplu, 99%), abordările bazate pe OTU au potențialul de a rezolva diversitatea la nivel de specie observată în intestinul uman. Menționăm, de asemenea, că, deși secvențierea 16S cu lungime întreagă poate fi optimă pentru analiza la nivel de specie, regiunile variabile extrem de informative (de exemplu, V1-V3) pot fi, de asemenea, adecvate în acest scop.,

Detectarea Bacteroides umane probe de scaun. a abundența relativă a genului Bacteroides în patru probe de scaun uman cuantificate folosind ampliconi V1-V9 (axa x) sau ampliconi V1–V3 (axa y). B abundența relativă a speciilor Bacteroides în aceleași patru probe. Abundența speciilor a fost cuantificată din secvențierea mWGS sau din Otu–urile V1-V3/V1-V9 generate la 99% identitate., Abundența este prezentată pentru speciile cele mai abundente, așa cum este cuantificată de mWGS (pentru estimările abundenței tuturor speciilor Bacteroides detectate de fiecare platformă, a se vedea tabelul suplimentar 5). profilele de substituție nucleotidică c generate prin alinierea tuturor secvențelor amplicon v1-V9 atribuite OTU unic identificat ca Bacteroides vulgatus. Profilurile sunt prezentate pentru cele două probe de scaun cu mare B. vulgatus abundență relativă (IronHorse și Scott). d Nucleotide substituție profile prezis de referință genomurile a două tipuri diferite de B. vulgatus tulpini ATCC 848239 și mpk40., În ambele c și d, substituții nucleotidice au fost identificate în raport cu o singură referință 16 gene pentru B. vulgatus ATCC 8482 (NCBI Gene ID 5304800). Panourile gri prezintă regiuni variabile definite de locurile de legare a grundurilor utilizate în mod obișnuit (tabelul suplimentar 1). Liniile punctate indică proporția așteptată de substituții de nucleotide, având în vedere că există șapte copii ale genei 16S în fiecare genom., Sursa de date sunt furnizate ca o Sursă fișier de Date
profitând de faptul că Bacteroides vulgatus a fost prezent la mare abundența relativă în două umane microbiomului intestinal probe, avem următoarea întrebat dacă intragenomic variație între 16 copii ale genei ar putea fi detectat in vivo. Am aliniat fiecare secvență de lungime completă clasificată ca aparținând B. vulgatus V1-V9 OTUs (Fig. 3b și date suplimentare 1) la o singură secvență genică B. vulgatus 16S reprezentativă. Am comparat apoi profilurile de substituție nucleotidică rezultate (Fig., 3c) cu profiluri prezise din două genomi de referință prezenți în baza de date NCBI Refseq19 (Fig. 3d).
majoritatea variației nucleotidelor prezente în nostru in vivo generat B. vulgatus OTU reflectat variația reală atribuite polimorfisme intragenomice. În schimb, variația probabil din cauza secvențiere erori apărut mici și mult sub minim ~14% frecvență care ar fi de așteptat dacă ar exista o singură B. vulgatus tulpina în fiecare probă, cu șapte 16 copii ale genei în genom (Fig. 3C, linii punctate).
deși nu știam numărul adevărat de B., tulpinile de vulgatus prezente în fiecare probă in vivo, s-a observat că ambele profiluri de substituție nucleotidică au o asemănare mai mare cu tulpina ATCC 8482 decât mpk. Variația a existat și la loci specifici care ar putea indica diferențe semnificative între genomii de referință in vivo și ATCC 8482. De exemplu, un singur polimorfism a fost detectat în regiunea V5 a ATCC 8482, care a fost prezent în trei copii 16S (43%). În primul eșantion in-vivo (Scott) acest polimorfism a fost prezent în 84% din Citire, în timp ce în al doilea (IronHorse) a fost prezent în 69% Din citire., Aceste numere corespund îndeaproape cu numerele așteptate dacă un polimorfism au fost prezente șase și cinci din șapte gene 16S, respectiv.
În concluzie, arătăm că secvențierea 16S de lungime completă a microbiomului intestinal uman poate rezolva cu precizie substituțiile cu un singur nucleotid care reflectă variația intragenomică între copiile genei 16S. Prezența unei astfel de variații indică faptul că secvențele 16S trebuie grupate pentru a reflecta unitățile taxonomice semnificative., Folosind OTUs grupate la 99% identitate, vom arăta că full-length 16S are potențialul de a oferi specii și chiar rezoluția taxonomică la nivel de tulpină. Analiza comunităților microbiene la aceste niveluri taxonomice promite să ofere o perspectivă foarte diferită de cea oferită de estimările abundenței la nivel de gen.
Intragenomic 16 polimorfisme sunt extrem de răspândită
Au demonstrat că este posibil pentru a rezolva intragenomic copia variante in vivo, avem următoarea căutat să stabilească măsura în care o astfel de copie variante apar în taxoni de obicei găsite în interiorul omului microbiomului intestinal., Am căutat în continuare să stabilim dacă astfel de profiluri pot fi utilizate în mod obișnuit pentru a distinge între tulpinile aceleiași specii.
am cultivat 381 de taxoni din microbiomul intestinal al persoanelor sănătoase descrise în Fig. 3, precum și de la alte persoane care participă la același studiu original20 (date suplimentare 2). Ulterior, am efectuat secvențierea genelor 16S de lungime completă pe izolate și citiri secvențiate aliniate pentru a identifica substituțiile nucleotidice caracteristice variantelor de copiere a genei 16S intragenomice.,clasificarea taxonomică a izolatelor a identificat 58 de specii presupuse (date suplimentare 2), în timp ce gruparea unei singure secvențe reprezentative pentru fiecare izolat la o similitudine de 99% a dus la 61 de Otu (cu între 1 și 73 de izolate atribuite fiecărui OTU). În total, 349 din 381 izolate secvențiate (54 Din 61 Otu) au avut unul sau mai multe SNP, indicând prezența polimorfismelor genei 16S, iar 205 profiluri SNP unice au fost identificate atunci când se ține cont de eroarea potențială de secvențiere (Fig. 4a și date suplimentare 2).

Intragenomic 16 polimorfismele genei umane microbiomului intestinal izolate. o locație a SNP-urilor prezente în genele 16S ale izolatelor bacteriene cultivate individual. Locațiile SNP au fost identificate prin eliminarea treptată a secvențelor de gene 16S de lungime întreagă generate pentru fiecare izolat individual. Axa X denotă poziția de-a lungul genei 16S. Axa Y denotă izolate individuale grupate pe baza filogeniei lor deduse. Regiunea albastru închis indică localizarea unui polimorfism., Pentru claritate, sunt prezentate maximum cinci izolate aparținând aceleiași specii. Pentru detalii privind profilurile de substituție nucleotidică pentru toate izolatele secvențiate, a se vedea datele suplimentare 2. b–d Exemple de nucleotide substituție profile arată tulpina-diferențele de nivel între izolatele identificate ca aparținând la trei specii bacteriene: b Shigella flexneri; c Bifidobacterium longum; d Collinsella aerofaciens. Pentru fiecare specie, sunt prezentate două profiluri de substituție nucleotidică izolate; cu toate acestea, exemple suplimentare pot fi găsite în datele suplimentare 2., Izolatele au fost identificate ca aparținând aceleiași specii dacă secvențele lor reprezentative au fost atribuite aceluiași OTU atunci când se grupează la identitatea secvenței de 99%. Identificarea taxonomică a fost efectuată utilizând BLAST pentru alinierea secvențelor reprezentative la baza de date BLAST NCBI 16S (vezi metode). Panourile gri prezintă regiuni variabile definite de locurile de legare a grundurilor utilizate în mod obișnuit (tabelul suplimentar 1). Liniile punctate indică proporția așteptată de substituții de nucleotide, având în vedere numărul de copii ale genei 16S prezis pentru fiecare genom., Sursa de date sunt furnizate ca o Sursă fișier de Date
în Special, compararea SNP profile pentru izolatele atribuit același OTU frecvent relevat diferențe în frecvența de Snp, care au fost sugestive de diferențe în intragenomic 16 copii ale genei strâns legate între taxoni. Exemple de profiluri de substituție diferite sunt prezentate pentru trei taxoni (Fig. 4B-d), care sunt sugestive pentru variația nivelului tulpinii comparabilă cu cea demonstrată în principiu pentru E. coli (Fig. 2b).,în concluzie, arătăm că mulți dintre membrii culturabili ai microbiomului intestinal uman posedă frecvent polimorfisme ale genelor 16S, care, atunci când sunt contabilizate corespunzător, au potențialul de a rezolva tulpini ale aceleiași specii.