koko 16S-geenin tarjoaa paremman taksonominen tarkkuus
~1500 bp 16S rRNA-geeni kuuluu yhdeksän muuttujan alueiden välissä koko hyvin säilynyt 16S-sekvenssin (Kuva. 1 A). Koko geenin sekvensointi toteutettiin alun perin Sanger-sekvensoinnilla., Tämä vaati Kloonaus geenejä, tuottaa, ja kokoaminen kaksi-kolme lukee klooni, ja tuottaa rajoitettu näytteenottosyvyys korkeilla kustannuksilla ja vaivaa. Tällä hetkellä kuitenkin valtaosa tutkimuksista järjestyksessä vain osa geeni, koska laajalti käytetty Illumina sekvensointi alusta (korkeampi suoritusteho, alentaa kustannuksia, vähentää vaivaa verrattuna Sanger) tuottaa lyhyitä sekvenssejä ( ≤ 300 emäkset)., Eri osa-alueiden geeni on siis kohdennettu, jotka vaihtelevat yhden muuttujan alueilla, kuten V4-tai V6, kolmen muuttujan alueilla, kuten V1–V3 tai V3–V5 (käytetty Human Microbiome Project yhdessä 454-sekvensointi platform9).
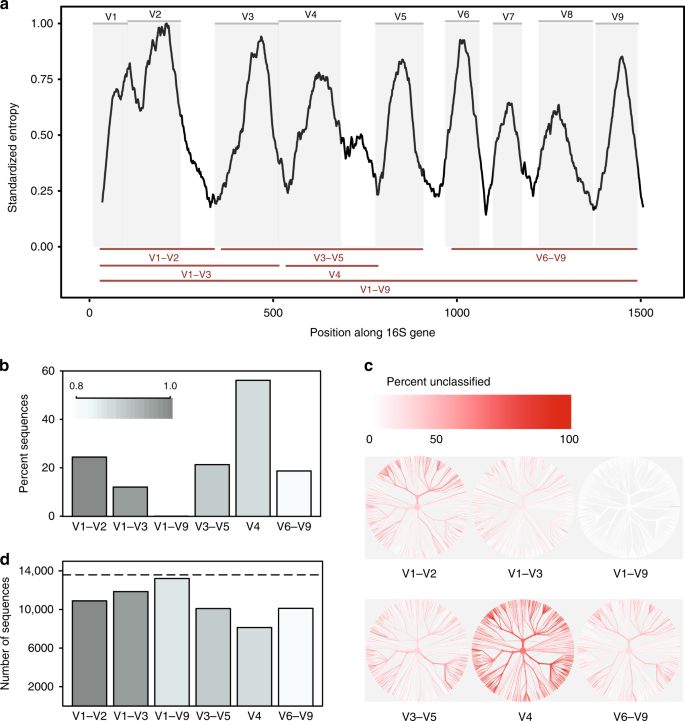
In-silico vertailu 16S rRNA-muuttuja alueilla. on Shannonin entropia koko 16S-geenin perustuva tasaus yhden edustajan järjestyksessä, jotta jokainen tunnettujen lajien läsnä Greengenes tietokantaan., Sekvenssit olivat kohdistettu yhden viittaus 16S-geenin Escherichia coli K-12 MG1655 (NCBI-Geenin ID 947777). Harmaat paneelit kuvaavat vaihtelevia alueita, jotka on määritelty yleisesti käytetyillä pohjustuskohteilla (Lisätaulukko 1). Tässä tutkimuksessa tarkasteltavat muuttuvat alueet esitetään punaisina viivoina (bottom). b osa-sekvenssit kunkin muuttujan alue, jota ei voitu tunnistaa lajin tarkkuudella luokittelussa jokainen sekvenssi vastaan viittaus tietokantaan, josta se on johdettu luottamusta kynnys 80% (RDP classifier)., C puut, jotka perustuvat in-silico-tietokannassa esiintyvien sekvenssien taksonomiaan. Sama puu on säädetty jokaiselle muuttuvalle alueelle. Väri kunkin haara heijastaa osuus sekvenssit kunkin haaran, jota ei voitu tunnistaa lajin tarkkuudella. d otusten lukumäärä, joka syntyy, kun kunkin muuttujan alueen sekvenssit ryhmittyvät 99%: n sekvenssien samankaltaisuuteen. Katkoviiva osoittaa useita ainutlaatuisia sekvenssejä (>1% erilaisia) alkuperäisessä tietokannassa., Lähde data tarjotaan Lähde Data file
Olemme sitä mieltä, että kohdistus osa-alueille edustaa historiallinen kompromissi, koska teknologia restrictions10. Tänään, sekä PacBio ja Oxford Nano-sekvensointi alustat pystyvät rutiininomaisesti tuottaa lukee yli 1500 bp ja korkea suoritusteho sekvensointi koko 16S-geenin on tulossa yhä yleisempiä., Ehdotamme sen vuoksi, että perustelut sille, että tämä kompromissi on tarkistettava ja me tehdään yksinkertainen in-silico-kokeilu osoittaa etu koko-pituus 16 sekvensointi yli kohdistus osa-alueille.
Me ladata joukko ei-tarpeeton (eli > 1% erilaisia), koko pituus 16S-sekvenssien julkisesta tietokannasta (Greengenes)., Hyödyntää sitä, että huomattava osa näistä sekvenssit otettu PCR-aluke-sitova sivustoja, me leikataan niitä tuottaa in-silico amplicons eri osa-alueilla, joka perustuu sijainnin PCR-alukkeita käytetään yleisesti microbiome tutkimukset (Fig. 1a ja Lisätaulukot 1-2)., Olettaen, että jokainen sekvenssi meidän ladannut tietokanta edustaa ainutlaatuinen laji, että voimme käyttää yhteisen luokituksen lähestymistapa (Ribosomi-Tietokanta-Hanke (RDP) classifier11) lasketaan taajuus, jolla in-silico amplicons jokaisesta osa-alueesta voi antaa tarkkoja, laji-level taksonominen luokittelu (käyttäen alkuperäisen tietokannan viite). Toisessa kokeessa, meillä on myös ryhmitelty in-silico amplicons tuottaa OTUs eri, yleisesti käytetty, sekvenssin samankaltaisuus kynnysarvot (97%, 98%, 99%).,
huomasimme, että osa-alueet erosivat toisistaan merkittävästi siinä, missä määrin he voisivat luotettavasti erottele koko-pituus 16S-sekvenssejä käytetään edustamaan lajia (Kuva. 1 B). V4-alueella suoritetaan pahin, 56% in-silico amplicons ei luottavaisesti vastaamaan niiden järjestys alkuperä tällä taksonominen taso. Sen sijaan, kun täysi-pituus järjestyksessä, jossa kaikki muuttujan alueilla oli käytetty, se oli mahdollista luokitella lähes kaikki sekvenssit kuin oikea laji (Supplementary Fig. 1 A)., Tietokantojen muuttaminen ja luokitusten luottamuskynnykset vaikuttivat in-silico ampliconien osuuteen, joka voitiin sovittaa tarkasti yhteen, mutta ei vaikuttanut vallitseviin suuntauksiin(täydentävä Kuva. 1 A, b).
Toiseksi, eri osa-alueiden osoitti puolueellisuudesta bakteeri-taksonien he pystyivät tunnistamaan (Kuva. 1c). Esimerkiksi V1–V2 alueella suoritetaan huonosti luokitella sekvenssejä, jotka kuuluvat pääjakso Proteobacteria, kun taas V3–V5 alueen heikkoa tällä luokitella sekvenssejä, jotka kuuluvat pääjakso Actinobacteria (Supplementary Fig. 2)., Vastaavanlaisia suuntauksia havaittiin sukutasolla taksoilla, joilla oli mahdollisesti lääketieteellistä merkitystä. Vaikka full V1–V9 alueen johdonmukaisesti tuottanut parhaat tulokset, V6–V9 alueella oli erityisesti paras osa-alueen luokitella sekvenssejä, jotka kuuluvat sukuihin, Clostridium-ja Staphylococcus, V3–V5 alueen tuottanut hyviä tuloksia Klebsiella, ja V1–V3 alueen tuottanut hyviä tuloksia Escherichia/Shigella (Supplementary Fig. 2 ja lähdetiedot).
Lopuksi, valinta osa-alueen vaikuttanut dramaattisesti määrä OTUs muodostuu, kun klusterointi in-silico amplicons luoda OTUs., Kun klusterointi on 99% identtisyys, kaikki osa-alueet ei luoda useita eri sekvenssejä läsnä alkuperäinen tietokanta; kuitenkin, V4-alueella taas suoritetaan pahin (Fig. 1d). Erityisesti suhteellinen määrä OTUs tuotettu kunkin osa-alueella ei ollut johdonmukainen eri identiteetti kynnysarvot (97%, 98%, 99%, Supplementary Fig. 3), mikä osoittaa, että käyttäytyminen klusterointi algoritmit voi olla vaikea ennustaa, milloin sisältämää tiedon määrää jaksotetaan alue on hyvin vaihteleva.,
lopuksi, kohdistus osa-alueille edustaa historiallinen kompromissi, joka oli riittävä tunnistaminen taksonien klo suvun tasolla tai yläpuolella. Kuitenkin, meidän yksinkertainen in-silico-koe osoittaa, että se ei ole perusteltua olettaa, että yhä hienompia klusterointi näiden osa-alueiden tulos on parantunut taksonominen tarkkuus tarpeen pohtia lajeja. Vaikka joillakin osa-alueilla (esim., V1–V3) antaa kohtuullinen lähentämisestä 16 monimuotoisuus, useimmat eivät kata riittävästi sekvenssin vaihtelu syrjiä läheisesti taksonien., Toteamme myös, että erotteleva polymorfismeja voidaan rajoittaa koskemaan tiettyjä muuttuvia alueita; sen vuoksi tietyt osa-alueet tulee paremmin erotteleva läheisesti jäsenet eräiden taksonien.
16S-geenikopiomuunnokset heijastavat kannanvaihtelua
16s-sekvenssien ryhmittelyä otukseen on historiallisesti palvellut kahta tarkoitusta. Ensinnäkin se on poistanut pieniä artifactual sekvenssivariantteja PCR-vahvistuksen ja sekvensointivirheiden vuoksi, kun se romahtaa sekvenssejä ryhmiksi. Toiseksi se on romahtanut laillisia sekvenssivariantteja, jotka ovat olemassa läheisen bakteeriperäisen taksan välillä., Vaikka jälkimmäinen ei ehkä aina ole toivottavaa, se järkeenkäypää, että et voi erottaa bakteeri-taksonien, joiden 16S-sekvenssien vaihdella nopeudella, joka on pienempi kuin virhe tapahtui erityisesti sekvensointialustamme.
viime aikoina CCS: n kehitys on parantanut huomattavasti pitkälukuisten sekvensointialustojen virhetasoja. Samalla laskennalliset menetelmät ovat mahdollistaneet laillisen vs. keinotekoisen sekvenssivaihtelun erottamisen toisistaan., Nämä teknologian ja menetelmien kehittyminen tarkoittaa tutkijat nyt on mahdollisuus suorittaa korkea-suoritusteho sekvensointi, joka voi tarkasti havaita yhden nukleotidin variantteja koko 16S-geenin.
Vaikka se on houkuttelevaa olettaa, että yhden nukleotidin vaihtoehdot voivat edustaa eri, läheisesti taksonien, me varoittaa siitä, että tämä on liian yksioikoinen tulkinta johtuu siitä, että monet bakteerien genomit sisältävät useita polymorfinen kopioita 16S gene12,13,14., Suoritimme PacBio CCS-sekvensoinnin 36 lajille bakteeri mock yhteisö(täydentävä Taulukko 3 ja täydentävä Kuva. 4) osoittaa, (i) että 16S-sekvenssin monet bakteerit vaihtelee välillä operons saman genomin ja (ii) että korkea suoritusteho sekvensointi on riittävän tarkka, jotta ratkaista nämä intragenomic eroja.
– Me ihmiset tietokoneella PacBio täyspitkä 16S-sekvenssejä on viitetietokanta, joka sisältää yhden edustajan 16S-sekvenssin jokainen jäsen meidän mock yhteisön ja käyttää linjaus tilastot arvioida paikkansapitävyyttä sekvensointi lähestymistapa., Vertaamalla ajokertojen määrä käytetään tuottamaan CCS-esiintyminen yhden nukleotidin vaihdot, lisäykset ja poistot ilmoitti, että kymmenen kulkee voisi minimoida nämä yhdistettynä virheet minimiin taajuus < 1.0% (vaikka se oli huomattavaa, että pienin saavutettavissa virhe vaihteli välillä sekvensointi toimii; Supplementary Fig. 5). Kuitenkin, teimme havaita yhteensattuma poisto virheitä sijainti homopolymer kulkee meidän viite sekvenssejä (täydentävä Kuva., 6), joka ei nukleotidi-erityisiä ja pahensi pituus sekvensoitiin homopolymeeri (Supplementary Fig. 7). Olemme sittemmin vahvistettu poistot sisällä Escherichia coli-bakteerin 16S-geenin käyttäen Illumina koko genomin haulikko (WGS) – sekvensointi, joka osoitti, että vain yksi poistot tapahtuvat PacBio sekvenssit oli aito (Supplementary Fig. 8).,
katsoo, että CCS-sekvensointi voi tuottaa 16S lukee matala taajuus korvaaminen virheet, me seuraavaksi perusteltu, että osan korvaaminen virheitä sisällä tarkasti linjassa lukee tulisi heijastaa vaihtelu johtuu 16S polymorfismeja lajin sisällä’ genome12. Esimerkiksi, lukee linjassa E. coli-kanta K-12 substr. MG1655 osoitti korvaaminen profiili, joka heijastuu juuri ennusti, että kohdistamalla kaikki seitsemän 16S-sekvenssien tiedetään olevan läsnä tässä genome15 (Fig. 2 A, c)., Olimme edelleen mahdollisuus vahvistaa stoikiometria näistä nukleotidin vaihdot määrällisesti vaihtelu on verrattain linjassa Illumina WGS lukee (Fig. 2b) ja osoitettava, että vastaava substituutioprofiili oli toistettavissa useissa jaksottamisajoissa (täydentävä Kuva. 9)., Rinnastuksia muihin viite sekvenssit meidän mock yhteisön näkyi samanlainen trendi runsas vaihdot lokalisoitu erityisiä pohja kantoja pitkin 16S-geenin, vaikka emme huomaa, että signaali-kohina-suhde kasvoi merkittävästi, kun 16S-geenin kysymys oli vähemmän kuin 100 tietokoneella lukee (Supplementary Fig. 10).

Polymorfismeja E. coli-bakteerin 16S rRNA geenisekvenssien. a kohdassa E esiintyvien substituutioiden sijainti ja esiintymistiheys., coli kanta K-12 MG1655 v1–V9 amplicons tuotettu meidän mock yhteisö ja sekvensoitu PacBio RS II alustalla. b-asema ja taajuus vaihtoja lukee syntyvät genomien sekvensointi eristetty E. coli-kanta K-12 MG1655 on Illumina MiSeq-alustan. Suurennettu alueiden näyttää kannat linjaus kaikkien seitsemän 16S geenit läsnä E. coli K-12 MG1655 viite genomin. Kaikkien SNP-vaiheistusten viitearvona käytetään 16S-sekvenssiä rrnd-operonista ( * * ). C E: n ennustettu nukleotidisubstituutioprofiili., coli K-12 MG1655 perustuu seitsemän 16S-geenisekvenssiä läsnä viite genomin. d ennustettu korvaaminen profiili E. coli O157: n Sakai perustuu kohdistamalla seitsemän 16S-geenin sekvenssit läsnä viite genomin. Harmaat paneelit kuvaavat vaihtelevia alueita, jotka on määritelty yleisesti käytetyillä pohjustuskohteilla (Lisätaulukko 1). Dashed linjat osoittavat odotettu osuus nukleotidisubstituutioita, koska on seitsemän 16S geeni kopioita kunkin genomin., Lähde data tarjotaan Lähde Data file
havainto, että pitkä-lue sekvensointi voi tunnistaa 16S polymorfismeja saman genomin on merkittäviä vaikutuksia. Ensinnäkin se osoittaa, että se ei ole perusteltua olettaa, että suurikapasiteettisten järjestyksessä lukee erilaiset yhden tai muutaman nukleotidin edustavat eri taxa6,16. Yhden perimän sisällä kaksi tai useampi 16S-sekvenssi voi olla identtisiä, kun taas toiset voivat olla ainutlaatuisia., Vastaavasti jotkut homologisia 16S loci voi säilyttää identtinen sekvenssi kahden läheistä sukua olevista kannoista, kun taas toiset voivat olla poikkesivat toisistaan yhden tai muutaman nukleotidin kantoja. Tässä yhteydessä minkä tahansa yhteisön tason tai taksonominen tulkinta 16S tietojen olisi mieluiten tilille siitä, että suhteellinen runsaus 16S-sekvenssejä, jotka johtuvat hyvin läheisesti taksonien heijastaa lineaarinen yhdistelmä (i) taajuus, jolla jokainen ainutlaatuinen sekvenssi on edustettuna koko genomien ja (ii) suhteellinen runsaus genomien kunkin taksonin.,
Toiseksi, vaikka intragenomic 16S-sekvenssin vaihtelu vaikeuttaa yhteisön tason analyysin, se on myös mahdollista lisätä valtaa 16S-geenin syrjiä läheisesti taksonien, koska sen avulla sekvenssi-pohjainen vertailu laajentaa useiden erilaisten loci. Esimerkiksi E. coli-kannan K-12 MG1655 erottamiseksi enterohemorragisesta kannasta O157 Sakai (Kuva. 2C, d)., Näin ollen olemme sitä mieltä, että, kun asianmukaisesti, useita polymorfinen 16S kopiot eivät haittaa voida jättää huomiotta, pikemminkin ne mahdollistavat 16S-geenin voidaan käyttää kanta-tason microbiome analyysi. Toteamme myös, että valta intragenomic 16S-sekvenssin vaihtelu syrjiä läheisesti taksonien on todennäköisesti vähenee, kun osittainen 16S-sekvenssejä käytetään. Esimerkiksi Kolibakteerikannat K-12 MG1655 (Kuva. 2C) alk. O157 Sakai (Kuva. 2D) esiintyy muuttuvilla alueilla V1, V2, V6 ja V9.,
16S-polymorfismit voidaan ratkaista in vivo
Mikrobiomiyhteisöt ovat usein monimutkaisia, olemassa erilaisissa biokemiallisissa ympäristöissä (esim.uloste, sylki, yskös jne.) ja sisältävät monia satoja ainutlaatuisia taksonien, joiden suhteellinen runsaus ulottuu laaja dynaaminen alue. Tämä monimutkaisuus ei ole hyvin edustettuna in-silico-tai mock-yhteisökokeissa. Siksi me suorittaa ylimääräinen kokeilu osoittaa, että sekvensointi koko 16S-geenin kun taas osuus intragenomic 16S SNPs voi ratkaista läheisesti bakteeri-taksonien in vivo.,
toteutimme PacBio CCS sekvensointi V1–V9 alueen neljän ihmisen jakkara näytteet kerätään terveillä vapaaehtoisilla aikuisilla. Vertailun vuoksi, me valmistimme V1–V3 alueella käyttäen Illumina MiSeq ja tarjotakseen vertailuarvon laji-level taksonominen kvantifiointi, me suoriteta metagenomic WGS (mWGS) sekvensointi käyttäen Illumina NextSeq. Arvioida, missä määrin kukin näistä sekvensointi lähestymistapoja voi ratkaista läheisesti taksonien, olemme keskittyneet Bacteroides-suvun., Sen lisäksi, että suku on runsas ihmisen suolistossa, se on hyvin monimuotoinen ja sisältää useita lajeja, jotka voivat vaikuttaa sekä hyviin että huonoihin vaikutuksiin17. Sitä on aiemmin käytetty myös mallitaksonina 16S-geenin hyödyllisyyden osoittamiseen korkean resoluution taksonomisessa analyysissä18.
Kun laskimme, Bacteroides runsaasti samaan sukuun tasolla, V1–V9 sekvensointi ja V1–V3 sekvensointi tuottanut vertailukelpoisia tuloksia., Molemmat lähestymistavat tunnistettu kaksi yksilöä, joilla on alhainen Bacteroides suhteellinen runsaus (~10-25%) ja kaksi henkilöä, joilla on korkea Bacteroides suhteellinen runsaus (~40-60%; Kuva. 3 A). Kuitenkin, laji-level kvantifioinnin kautta mWGS sekvensointi paljasti paljon suurempi monimuotoisuus, eri Bacteroides-lajit hallitseva suolistossa kunkin yksittäisen (Fig. 3b ja lisätiedot 1). Kun OTUs oli 99-prosenttisesti tunnistettavissa, sekä v1–V9 että V1-V3-sekvensointi pystyivät heijastamaan tätä lajitason vaihtelua (kuva., 3b), lukuun ottamatta, että V1–V3 sekvensointi ei havaita, Bacteroides intestinalis, joka oli runsas yksi neljästä microbiome ihmisen suolistossa näytteitä. Näiden tulosten perusteella voimme päätellä, että, kun sitä käytetään yhdessä asianmukaisen identiteetin kynnys (esim., 99%), OTU-perustuvat lähestymistavat ovat mahdollisia ratkaista laji-level diversity havaittu ihmisen suolistossa. Voimme edelleen todeta, että vaikka täysi-pituus 16 sekvensointi voi olla optimaalinen laji-tason analyysi, erittäin informatiivinen muuttuja-alueiden (esim., V1–V3), voi myös olla sopiva tähän tarkoitukseen.,

Havaita Bacteroides ihmisen jakkara näytteitä. a suhteellinen runsaus Bacteroides-suvun neljässä ihmisen jakkara näytteitä määrittää käyttämällä joko V1–V9 amplicons (x-akseli) tai V1–V3 amplicons (y-akseli). b Bakteroidien lajien suhteellinen runsaus samoissa neljässä näytteessä. Lajin runsaus oli kvantifioida mWGS sekvensointi tai V1–V3/V1–V9 OTUs syntyy 99% identiteettiä., Runsaus on esitetty runsaimmat lajit kuten kvantifioidaan mWGS (runsaus arviot kaikista Bacteroides-lajit havaita kunkin alustan, ks. Täydentävä Taulukko 5). c Nukleotidin korvaaminen profiilit syntyy kohdistamalla kaikki V1–V9 amplicon sekvenssit määritetty yhden OTU tunnistettu Bacteroides vulgatus. Profiilit on esitetty kahdesta ulostenäytteestä, joissa on korkea B. vulgatus suhteellinen runsaus (IronHorse ja Scott). d Nukleotidin korvaaminen profiilit ennustaa viite genomeja kahden eri B. vulgatus ATCC kantoja 848239 ja mpk40., Sekä c-ja d -, nukleotidi-substituutioita havaittiin suhteessa yksi viittaus 16S-geenin B. vulgatus ATCC 8482 (NCBI-Geenin ID 5304800). Harmaat paneelit kuvaavat vaihtelevia alueita, jotka on määritelty yleisesti käytetyillä pohjustuskohteilla (Lisätaulukko 1). Dashed linjat osoittavat odotettu osuus nukleotidisubstituutioita, koska on seitsemän 16S geeni kopioita kunkin genomin., Lähde data tarjotaan Lähde Data file
hyödyntää sitä, että Bacteroides vulgatus oli läsnä korkea suhteellinen runsaus kahden ihmisen gut microbiome näytteitä, me seuraavaksi kysyttiin, onko intragenomic vaihtelu 16S-geenin kopiota ei havaittu in vivo. Me ihmiset tietokoneella jokainen täysi-pituus järjestyksessä luokiteltu kuuluviksi B. vulgatus V1–V9 OTUs (Fig. 3b ja lisätiedot 1) yhteen edustavaan B. vulgatus 16S-geenisarjaan. Tämän jälkeen vertailimme tuloksena syntyneitä nukleotidisubstituutioprofiileja (Kuva., 3c) profiileilla, jotka on ennustettu kahdesta NCBI: n RefSeq-tietokannassa19 (Kuva. 3D).
suurin osa nukleotidin vaihtelua läsnä meidän in vivo syntyy B. vulgatus OTU näkyy todellinen vaihtelu johtuu intragenomic polymorfismeja. Sen sijaan vaihtelu johtuu todennäköisesti sekvensointi virheitä ilmestyi alhainen ja selvästi alle minimaalinen ~14% taajuus, joka olisi odotettavissa, jos siellä oli yksi B. vulgatus kanta kunkin näytteen seitsemän 16S-geenin kopiota sen genomin (Fig. 3C, dashed linjat).
vaikka emme tienneet B: n todellista lukua., vulgatus kantoja läsnä jokaisessa in vivo-näyte, se oli huomattavaa, että molemmat nukleotidin korvaaminen profiilit kantoi lähempänä yhdennäköisyys kanta ATCC 8482 kuin mpk. Vaihtelua on myös olemassa erityisiä loci, joka voisi mahdollisesti osoittaa mielekkäitä eroja in vivo-ja ATCC 8482 viite genomeja. Esimerkiksi ATCC 8482: n V5-alueella havaittiin yksi polymorfismi, jota oli kolme 16S-kopiota (43%). Ensimmäinen in vivo-näyte (Scott) tämä polymorfismi oli läsnä 84% lukee, katsoo, että toinen (IronHorse) se oli läsnä 69% lukee., Nämä numerot vastaavat tarkasti numeroita odotettavissa, jos polymorfismi oli läsnä kuusi ja viisi seitsemästä 16S geenit, vastaavasti.
lopuksi, me osoittavat, että koko-pituus 16 sekvensointi ihmisen gut microbiome voi tarkasti ratkaista yhden nukleotidin vaihdot, jotka heijastavat intragenomic vaihtelu 16S-geenin kopioita. Tällaisen vaihtelun esiintyminen osoittaa, että 16S-sekvenssit on ryhmiteltävä mielekkäiden taksonomisten yksiköiden mukaisiksi., Käyttämällä OTUs clustered 99% identiteetti, osoitamme, että kokopitkä 16S on mahdollisuus tarjota lajeja ja jopa kanta-tason taksonominen resoluutio. Analyysi mikrobien yhteisöjen nämä taksonomiset tasot lupaa tarjota hyvin erilaisen näkökulman yksi tarjoamia suku-tasolla, runsaasti arvioita.
Intragenomic 16S polymorfismit ovat erittäin yleisiä
Ottaa osoittanut, että se on mahdollista ratkaista intragenomic kopioi variantteja in vivo, me seuraavaksi pyrittiin selvittämään, missä määrin tällainen kopio vaihtoehdot näkyvät taksonien yleisesti todettu sisällä microbiome ihmisen suolistossa., Pyrimme edelleen selvittämään, voidaanko tällaisia profiileja käyttää rutiininomaisesti erottamaan saman lajin kannat.
– Meillä viljellyt 381 taksonien from the gut microbiome terveitä yksilöitä kuvattu Kuva. 3 sekä muilta samaan alkuperäiseen tutkimukseen20 osallistuvilta henkilöiltä (lisätiedot 2). Tämän jälkeen suoritetaan koko-pituus 16S-geenin sekvensointi on eristää ja tietokoneella sekvensoitiin lukee tunnistaa nukleotidin vaihdot ominaisuus intragenomic 16S-geenin kopio variantteja.,
Taksonominen luokittelu isolaatteja tunnistetaan 58 oletetun lajin (Täydentävä tieto 2), kun taas klustereiden yksi edustaja järjestyksessä kunkin eristää 99% samankaltaisuus johti 61 OTUs (välillä 1 ja 73 eristää kullekin OTU). Yhteensä 349 381 sekvensoitiin isolaatit (54 61 OTUs) oli yksi tai useampi SNP, joka osoittaa läsnäolo 16S-geenin polymorfismit, ja 205 ainutlaatuinen SNP-profiilit havaittiin, kun osuus potentiaalia sekvensointi virhe (Kuva. 4a ja lisätiedot 2).

Intragenomic 16S-geenin polymorfismit ihmisen gut microbiome-isolaatteja. sijainti SNPs läsnä 16S geenejä yksilöllisesti viljelty bakteeri isolaattien. SNP: n sijainnit tunnistettiin vaiheistamalla kullekin yksittäiselle isolaatille syntyneitä täyspitkiä 16S-geenisekvenssejä. X-akseli tarkoittaa sijaintia 16S-geenin varrella. Y-akseli tarkoittaa yksittäisiä isolaatteja, jotka on koottu niiden päätellyn fylogenian perusteella. Tummansininen alue ilmaisee polymorfismin sijainnin., Selkeyden vuoksi esitetään enintään viisi samaan lajiin kuuluvaa isolaattia. Lisätietoa nukleotidisubstituutioprofiileista kaikkien sekvensoitujen isolaattien osalta, ks.lisätiedot 2. b–d-Esimerkkejä nukleotidin korvaaminen profiilit osoittaa kanta-tasolla erot isolaatteja tunnistetaan kuuluvaksi kolme bakteeri laji: b Shigella flexneri; c Bifidobacterium longum; d Collinsella aerofaciens. Kustakin lajista esitetään kaksi isolaattinukleotidisubstituutioprofiilia; lisäesimerkkejä löytyy kuitenkin Lisäaineistosta 2., Isolaattia tunnistettiin, jotka kuuluvat samaan lajiin, jos heidän edustajansa sekvenssit olivat kohdistettu samalle OTU, kun klusterointi on 99% identtisyys. Taksonominen tunnistaminen suoritettiin BLAST-menetelmällä edustavien sekvenssien sovittamiseksi NCBI 16s BLAST-tietokantaan (KS.menetelmät). Harmaat paneelit kuvaavat vaihtelevia alueita, jotka on määritelty yleisesti käytetyillä pohjustuskohteilla (Lisätaulukko 1). Katkoviivat osoittavat odotettua osuus nukleotidin vaihdot, koska määrä 16S-geenin kopioita ennusti kunkin genomin., Lähde data tarjotaan Lähde Data file
Erityisesti, vertaamalla SNP-profiileja eristää määritetty sama OTU usein paljasti eroja taajuus SNPs, jotka olivat viittaavia eroja intragenomic 16S-geenin kopioiden välillä läheisesti taksonien. Esimerkkejä erilaisista korvausprofiileista esitetään kolmesta taksasta (Kuva. 4b-d), jotka viittaavat kannanvaihteluun, joka on verrattavissa E. coli-bakteerin (Fig. 2 B).,
lopuksi, me osoittavat, että monet viljeltävien jäseniä ihmisen gut microbiome usein hallussaan 16S-geenin polymorfismeja, jotka, kun oikein laskettu, on mahdollista ratkaista kantoja samaa lajia.