RNA-sekvensointi (Wang 2009) on nopeasti korvaamassa geenien ilmentyminen microarray monissa labs. RNA-seq avulla voit määrittää, löytää ja profiloida RNAs. Tätä tekniikkaa varten mRNA (ja muut RNAs) muunnetaan ensin cDNA: ksi. Tämän jälkeen cDNA: ta käytetään seuraavan sukupolven sekvensointikirjaston valmistelun syötteenä. Tässä artikkelissa, annan lyhyen tarkastelun RNA-seq ja esitellä tärkeimmät menetelmät käytetään tänään.
miksi RNA-Seq on ”parempi” kuin mikrorakenteet?,
On olemassa useita etuja RNA-seq on yli microarray:
Kanssa RNA-seq, voit kuulustella enemmän kuin vain differentiaali-geenin ilmentyminen. Vaikka on olemassa microarray saatavilla eksonin-tasolla ja microRNA-analyysi, useimmat käyttäjät ovat edelleen kiinnostuneita perus, varmaan 3′ puolueellinen, differentiaali-geenin ilmentyminen. Kanssa RNA-seq, voit katsoa koodaus ja non-coding RNA, klo liittämiseen ja alleelia erityinen ilmaisu, ja mahdollisesti pian täyspitkän cDNA-sekvenssit, poistaa tarpeen päätellä tai koota isoentsyymien.,
mikrorakenteet ovat myös puolueellisia, koska meidän on päätettävä, mitä sisältöä sijoittaa matriisiin. Koska RNA-seq ei käytä koettimia tai alukkeita, tiedot kärsivät paljon pienempi harhat (vaikka en tarkoita sanoa, RNA-seq on ei mitään).
RNA-seq tarjoaa digitaalisen datan muodossa tietokoneella lukea-laskee, mikä erittäin laaja dynaaminen alue, parantaa herkkyyttä havaita harvinaisia selostukset.
Se on myös erittäin kilpailukykyinen hinnaltaan, jotta microarray, kuten tänään, välillä 6-30 näytteitä voidaan kanavoida yhden Illumina sekvensointi lane.,
Lopuksi, voit uudelleenanalysointi RNA-seq-datajoukon, mitä enemmän tietoa transcriptome tulee saataville. Jos paperi on julkaistu osoittaa mielenkiintoinen liitos-variantti vastaavan järjestelmän yksi työ, sitten haluat ehkä mennä takaisin ja katsoa, että liitos teidän näytteitä; ja sinulla olisi jo tiedot tehdä niin.
miten RNA-Seq vaikuttaa?
RNA-seq-kokeen suorittamiseen on monia menetelmiä. Itse asiassa, tekniikat kehittyvät niin nopeasti, se voi olla vaikea päättää, mitä käyttää., Perus valinta on välillä 1) random-pohjamaalattu cDNA synteesi kaksijuosteista cDNA tai 2) RNA-ligaatio-menetelmän (uudelleen ja verrata Levin 2010). Useimmat ihmiset käyttävät ensimmäistä menetelmää ja sitten on tehtävä lisävalinta lohkokohtaisen protokollan ja sellaisen välillä, joka ei ole. Eniten laboratoriossani käytetty menetelmä on Illuminan TruSeq RNA-seq, joka on satunnaistettu cDNA-synteesin ei-strand-spesifinen protokolla.
kun sinulla on sekvensointikirjasto, se jaksotetaan tiettyyn syvyyteen, joka riippuu siitä, mitä haluat tehdä tiedoilla., Nämä tulokset ovat linjassa genomin tai transcriptome ja lasketaan määrittää ero geenien ilmentyminen tai analysoida tarkemmin määritellä, liittämiseen ja-isoentsyymin ilme. Useimmat ihmiset sekvensoivat RNA: ta pariläpäisellä 50-100bp-menetelmällä. Poikkeuksena on microRNA-sekvensointi, koska se edellyttää useimmissa tapauksissa vain yhden pään 36bp-sekvensointia.
Meidän RNA-Seq-Menetelmä
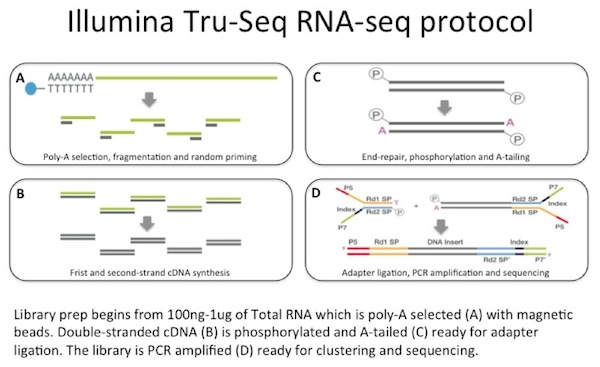
käytämme välillä 100 ng 1 µg kokonais-RNA: n tulona mRNA capture oligo-dT päällystetty magneettinen helmiä. MRNA on sirpaleinen, minkä jälkeen suoritetaan satunnaistettu cDNA-synteesi., Tuloksena double strand cDNA käytetään input-standardin Illumina library prep joka sisältää pää-korjaus, sovitin sitomiseen ja PCR-monistus antaa sinulle library valmis sekvensointi.
Miksi Vaivautua Strand Tietoa?
sense-transkriptiosta ja sen biologisesta merkityksestä on keskusteltu paljon. Jos olet kiinnostunut yksinkertainen differentiaali-geenin ilmentyminen, sitten lohkon tietoja ei lisätä paljon kokeilun, mutta tekevät pöytäkirjan enemmän monimutkainen., Ottaa sanoi, että, voit suorittaa laajimmin hyväksytty menetelmä ilman liikaa ylimääräistä vaivaa. Voit tehdä tämän käyttämällä 2. Strandin cDNA-synteesin aikana urasiilia inkorporaationa tymiinin sijaan. Seuraa Illumina-kirjastoa normaalisti, mutta adapterin ligaation jälkeen ja ennen PCR-vahvistusta lisää urasiili-DNA-glykosylaasi 2ndstrandin hajottamiseksi. Tämä johtaa kaikki lukee alkaen samassa suuntauksessa, joten voit määrittää, mikä strand oli litteroitu teidän näyte.
mitä RNA-Seq: lla voi oikeasti tehdä?,
RNA-seq on voimakas ja monipuolinen työkalu, jota on julkaistu laajasti viime vuosina. Olen poiminut pari suosikkiani (osa tekemistäni töistä johtamassani ydinlaitoksessa) havainnollistamaan, mitä RNA-sekvensoinnilla voi tehdä.
- Jabbari, et al. RNA-seq: ta käytettiin psoriaasin tutkimiseen ja uusien geenien löytämiseen funktionaalista analyysia varten. He vertasivat RNA-seq-tietojaan julkaistuihin matriisitutkimuksiin ja löysivät 1700 uutta hakijaa. QPCR vahvisti ne, ja vertailu psoriasiksen toiminnallisiin tietokantoihin tuki niiden roolia patogeneesissä.
- Kutter, et al., käytetty RNA-seq tutkimuksessa tarkastellaan säilyttämistä RNA-Polymeraasi III sitova nisäkkäillä vahvistaa geenien käytössä Pol III, joka määritetään ChIP-seq.
- Mercer, et al. yhdistetty RNA-seq ja mikroarraypohjainen talteenotto harvinaisten transkriptioiden tunnistamiseksi ja luonnehtimiseksi, jotka ovat normaalisti havaitsematta. Suunnattu selostukset lisääntynyt järjestyksessä lukea runsaasti alkaen 0.21% pre-capture 80% post kaapata., He löysivät yli 200 aiemmin unannotated isoentsyymien kautta, sillä lähes 50 proteiini-koodaus loci, mukaan lukien uusi vaihtoehto isomuodon TP53, joka on erittäin hyvin tunnettu geeni. Tämä viittaa siihen, että perimässä ja transcriptomessa on vielä paljon monimutkaisuutta ratkaistavana.
yhteenvetona, RNA-seq on edelleen kehittyvä työkalu, mutta on parempi on useimmissa tapauksissa microarray. Se on herkempi, vankempi ja voi olla kustannustehokkaampi. Mitä RNA-seq-projekteja suunnittelet nyt projektillesi?,
Jabbari et al: psoriasiksen Transcriptionaalinen profilointi RNA-seq: n avulla paljastaa aiemmin tunnistamattomia differentiaalisesti ilmaistuja geenejä. Journal of Investigative Dermatology 2011.
– Kutter ym: Pol III sitova kuusi nisäkkäitä osoittaa säilyttämistä keskuudessa aminohappo isotypes huolimatta eroja tRNA geenit. Luontogenetiikka 2011.
Levin et al: strand-spesifisten RNA-sekvensointimenetelmien kattava vertaileva analyysi. Luontomenetelmät 2010.
Mercer et al: Kohdennettu RNA-sekvensointi paljastaa ihmisen transkriptomin syvän monimutkaisuuden. Luontobioteknologia 2012.,
Wang et al: RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009.
julkaistiin alun perin 25.toukokuuta 2012. Päivitetty ja tarkistettu 16. elokuuta 2015.

onko tämä auttanut sinua? Jaa sitten verkkosi kanssa.