Il sequenziamento dell’RNA (Wang 2009) sta rapidamente sostituendo i microarray di espressione genica in molti laboratori. RNA-seq consente di quantificare, scoprire e profilo RNA. Per questa tecnica, l’mRNA (e altri RNA) vengono prima convertiti in cDNA. Il cDNA viene quindi utilizzato come input per la preparazione di una libreria di sequenziamento di nuova generazione. In questo articolo, darò una breve rassegna di RNA-seq e introdurre i principali metodi utilizzati oggi.
Perché RNA-Seq è “migliore” dei microarray?,
Ci sono diversi vantaggi che RNA-seq ha rispetto ai microarray:
Con RNA-seq è possibile interrogare più di una semplice espressione genica differenziale. Anche se ci sono microarray disponibili per l’analisi a livello di esone e microRNA, la maggior parte degli utenti sono ancora interessati a base, probabilmente 3’ polarizzato, espressione genica differenziale. Con RNA-seq si può guardare a codifica e non codifica RNA, a splicing e allele espressione specifica, e forse presto a full-length sequenze cDNA, eliminando la necessità di dedurre o assemblare isoforme.,
Anche i microarray sono di parte, poiché dobbiamo decidere quale contenuto inserire nell’array. Poiché RNA-seq non utilizza sonde o primer, i dati soffrono di pregiudizi molto più bassi (anche se non intendo dire che RNA-seq non ne abbia nessuno).
RNA-seq fornisce dati digitali sotto forma di conteggi allineati, risultando in una gamma dinamica molto ampia, migliorando la sensibilità di rilevamento per trascrizioni rare.
È anche molto costo-competitivo per microarray, come oggi, tra 6-30 campioni possono essere multiplexati in un unico Illumina sequenziamento corsia.,
Infine, è possibile rianalizzare un set di dati RNA-seq man mano che diventano disponibili ulteriori informazioni sul trascrittoma. Se viene pubblicato un documento che mostra un’interessante variante di splice in un sistema simile a quello su cui lavori, potresti voler tornare indietro e guardare quello splicing nei tuoi campioni; e avresti già i dati per farlo.
Come agisce RNA-Seq?
Esistono molti metodi per eseguire un esperimento RNA-seq. In effetti, le tecniche si stanno evolvendo così rapidamente che può essere difficile decidere quale usare., Una scelta di base è tra 1) sintesi di cDNA con primer casuale da cDNA a doppio filamento o 2) Metodi di legatura dell’RNA (esaminati e confrontati in Levin 2010). La maggior parte delle persone utilizza il primo metodo e quindi ha bisogno di fare un’ulteriore scelta tra un protocollo specifico del filo e uno che non lo è. Il metodo più utilizzato nel mio laboratorio è Truseq RNA-seq di Illumina, che è un protocollo di sintesi non-strand-specifico di cDNA con primer casuale.
Una volta che hai una libreria di sequenziamento, viene sequenziata a una profondità specificata, che dipende da cosa vuoi fare con i dati., Queste letture sono allineate al genoma o al trascrittoma e vengono contate per determinare l’espressione genica differenziale o ulteriormente analizzate per determinare l’espressione di splicing e isoforma. La maggior parte delle persone sta sequenziando l’RNA usando metodi 50-100bp accoppiati. L’eccezione è il sequenziamento microRNA, in quanto ciò richiede solo sequenziamento 36bp single-end nella maggior parte dei casi.
Il nostro metodo RNA-Seq
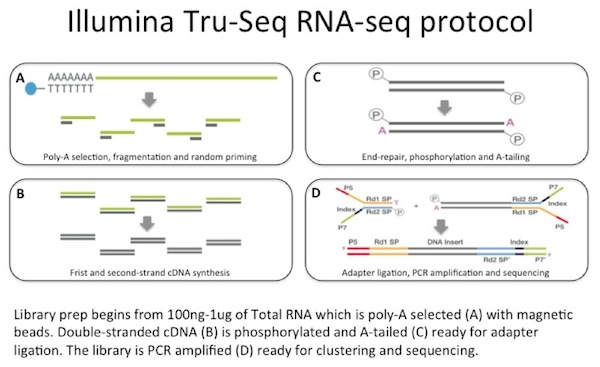
Usiamo tra 100 ng e 1 µg di RNA totale come input per una cattura di mRNA con perline magnetiche rivestite con oligo-dT. L’mRNA è frammentato e quindi viene eseguita una sintesi di cDNA innescata a caso., Il cDNA a doppio filamento risultante viene utilizzato come input per una preparazione della libreria Illumina standard che include la riparazione finale, la legatura dell’adattatore e l’amplificazione PCR per fornire una libreria pronta per il sequenziamento.
Perché preoccuparsi di informazioni Strand?
C’è stata molta discussione sulla trascrizione anti-senso e sulla sua rilevanza biologica. Se sei interessato alla semplice espressione genica differenziale, le informazioni sul filo non aggiungeranno molto al tuo esperimento, ma renderanno il tuo protocollo più complesso., Detto questo, è possibile eseguire il metodo più ampiamente adottato senza troppi sforzi extra. Per fare questo, durante la sintesi del cDNA del secondo filamento, usi l’uracile per incorporazione invece della timina. Seguire la preparazione della libreria Illumina normalmente, ma dopo la legatura dell’adattatore e prima dell’amplificazione della PCR aggiungere uracil-DNA glicosilasi per degradare il 2ndstrand. Ciò si traduce in tutte le letture che iniziano con lo stesso orientamento in modo da poter determinare quale filo veniva trascritto nel campione.
Che cosa si può effettivamente fare con RNA-Seq?,
RNA-seq è uno strumento potente e versatile pubblicato ampiamente negli ultimi anni. Ho scelto un paio dei miei preferiti (alcuni dal lavoro svolto nella struttura principale che gestisco) per illustrare cosa si può fare con il sequenziamento dell’RNA.
- Jabbari, et al. usato RNA-seq per studiare la psoriasi e trovare nuovi geni per l’analisi funzionale. Hanno confrontato i loro dati RNA-seq con studi di array pubblicati e hanno trovato 1700 nuovi candidati. Questi sono stati convalidati da qPCR e il confronto con i database funzionali per la psoriasi ha supportato il loro ruolo nella patogenesi.
- Kutter, et al., utilizzato RNA-seq in uno studio che esamina la conservazione del legame della RNA polimerasi III nei mammiferi per convalidare l’espressione dei geni occupati da Pol III come analizzato da ChIP-seq.
- Mercer, et al. RNA-seq combinato e cattura basata su microarray per identificare e caratterizzare trascritti rari, che normalmente non sono rilevabili. Le trascrizioni mirate sono aumentate in abbondanza di lettura in sequenza da 0.21% pre-capture a 80% post capture., Hanno trovato più di 200 isoforme precedentemente non annotate per quasi 50 loci codificanti proteine, inclusa una nuova isoforma alternativa di TP53, che è un gene molto ben caratterizzato. Ciò suggerisce che c’è ancora molta complessità nel genoma e nel trascrittoma da risolvere.
In sintesi, RNA-seq è ancora uno strumento in evoluzione, ma è preferibile nella maggior parte dei casi ai microarray. È più sensibile, più robusto e può essere più conveniente. Quali progetti RNA-seq stai pianificando per il tuo progetto?,
Jabbari et al: Il profilo trascrizionale della psoriasi utilizzando RNA-seq rivela geni precedentemente non identificati espressi in modo differenziale. Rivista di Dermatologia investigativa 2011.
Kutter et al: Il legame Pol III in sei mammiferi mostra la conservazione tra gli isotipi di aminoacidi nonostante la divergenza tra i geni tRNA. Nature Genetics 2011.
Levin et al: Analisi comparativa completa dei metodi di sequenziamento del filamento specifico di RNA. Metodi della natura 2010.
Mercer et al: Il sequenziamento mirato dell’RNA rivela la profonda complessità del trascrittoma umano. Natura Biotecnologia 2012.,
Wang et al: RNA-Seq: uno strumento rivoluzionario per la trascrittomica. Nat Rev Genet 2009.
Originariamente pubblicato il 25 maggio 2012. Aggiornato e rivisto il 16 agosto 2015.

Questo ti ha aiutato? Quindi si prega di condividere con la rete.