Il gene 16S completo fornisce una migliore risoluzione tassonomica
Il gene rRNA ~ 1500 bp 16S comprende nove regioni variabili intervallate 1 bis). Sequenziamento l’intero gene è stato originariamente realizzato da Sanger sequenziamento., Ciò richiedeva la clonazione di geni, la generazione e l’assemblaggio di due o tre letture per clone e la produzione di una profondità di campionamento limitata a costi e sforzi elevati. Attualmente, tuttavia, la stragrande maggioranza degli studi sequenziano solo una parte del gene, perché la piattaforma di sequenziamento Illumina ampiamente utilizzata (throughput più elevato, costo inferiore, sforzo ridotto rispetto a Sanger) produce sequenze brevi (≤300 basi)., Sono quindi prese di mira diverse sottoregioni del gene, che vanno da singole regioni variabili, come V4 o V6, a tre regioni variabili, come V1-V3 o V3–V5 (utilizzate nel progetto Microbiome umano in combinazione con la piattaforma di sequenziamento 4549).
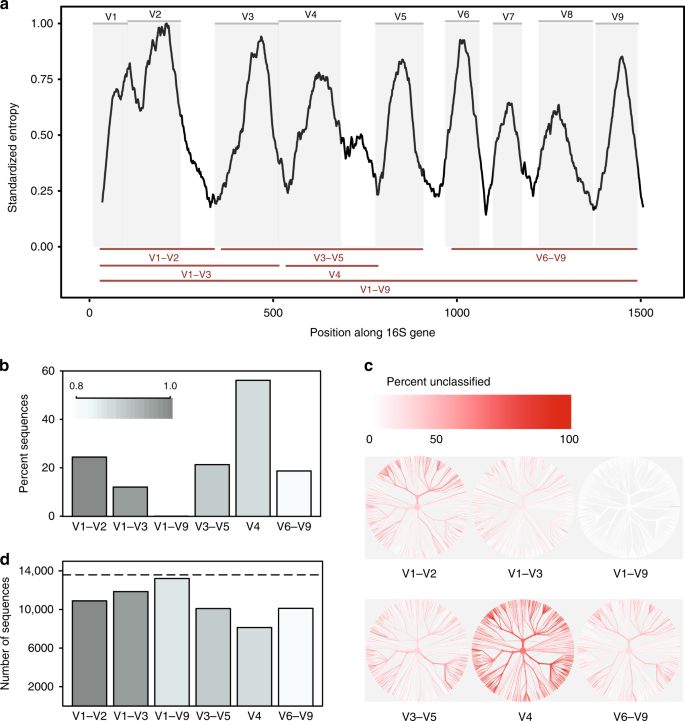
Confronto in-silico delle regioni variabili rRNA 16S. un’entropia di Shannon attraverso il gene 16S basata sull’allineamento di una singola sequenza rappresentativa per ogni specie nota presente nel database Greengenes., Le sequenze sono state allineate contro un singolo gene di riferimento 16S per Escherichia coli K-12 MG1655 (gene NCBI ID 947777). I pannelli grigi raffigurano regioni variabili definite da siti di legatura del primer comunemente usati (Tabella supplementare 1). Le regioni variabili considerate in questo studio sono mostrate come linee rosse (in basso). b Proporzione di sequenze per ogni regione variabile che non è stato possibile identificare a livello di specie quando si classifica ciascuna sequenza rispetto alla banca dati di riferimento da cui è stata ricavata a una soglia di confidenza dell ‘ 80% (classificatore RDP)., c Alberi basati sulla tassonomia delle sequenze presenti nel database in-silico. Lo stesso albero viene fornito per ogni regione variabile. Il colore di ogni ramo riflette la proporzione di sequenze all’interno di ciascun clade che non è stato possibile identificare a livello di specie. d Il numero di OTU creati durante il clustering di sequenze per ciascuna regione variabile con una somiglianza di sequenza del 99%. La riga tratteggiata indica il numero di sequenze univoche (>1% diverso) nel database originale., I dati di origine sono forniti come file di dati di origine
Sosteniamo che il targeting delle sub-regioni rappresenta un compromesso storico, a causa delle restrizioni tecnologiche10. Oggi, sia le piattaforme di sequenziamento di nanopori PacBio che Oxford sono in grado di produrre regolarmente letture superiori a 1500 bp e il sequenziamento ad alto throughput del gene 16S completo sta diventando sempre più diffuso., Pertanto suggeriamo che la giustificazione di questo compromesso debba essere rivisitata e abbiamo eseguito un semplice esperimento in-silico per dimostrare il vantaggio del sequenziamento 16S a lunghezza intera rispetto al targeting delle sub-regioni.
Abbiamo scaricato una serie di sequenze 16S non ridondanti (cioè> 1% diverse), a lunghezza intera da un database pubblico (Greengenes)., Approfittando del fatto che una parte sostanziale di queste sequenze incorporava siti di legame del primer PCR, li abbiamo tagliati per generare ampliconi in-silico per diverse sottoregioni, in base alla posizione dei primer PCR comunemente usati negli studi sul microbioma (Fig. 1a e Tabelle supplementari 1-2)., Supponendo che ogni sequenza nel nostro database scaricato rappresentasse una specie unica, abbiamo quindi utilizzato un approccio di classificazione comune (il classificatore Ribosome Database Project (RDP) 11) per calcolare la frequenza con cui gli ampliconi in-silico per ogni sottoregione potrebbero fornire una classificazione tassonomica accurata a livello di specie (utilizzando il database originale come riferimento). In un secondo esperimento, abbiamo anche raggruppato i nostri ampliconi in-silico per generare OTU a soglie di somiglianza di sequenza diverse, comunemente usate(97%, 98%, 99%).,
Abbiamo scoperto che le sottoregioni differivano sostanzialmente nella misura in cui potevano tranquillamente discriminare tra le sequenze 16S a lunghezza intera utilizzate per rappresentare le specie (Fig. 1 ter). La regione V4 ha avuto risultati peggiori, con il 56% degli ampliconi in-silico che non riescono a corrispondere con sicurezza alla loro sequenza di origine a questo livello tassonomico. Al contrario, quando è stata utilizzata una sequenza a lunghezza intera con tutte le regioni variabili, è stato possibile classificare quasi tutte le sequenze come specie corrette (Fig. 1 bis)., La modifica delle basi di dati e delle soglie di confidenza della classificazione ha influito sulla percentuale di ampliconi in-silico che potevano essere accuratamente abbinati, ma non ha influenzato le tendenze prevalenti (Fig. 1 bis, lettera b).
In secondo luogo, diverse sub-regioni hanno mostrato bias nei taxa batterici che sono stati in grado di identificare (Fig. 1c). Ad esempio, la regione V1–V2 si è comportata male nella classificazione delle sequenze appartenenti al phylum Proteobacteria, mentre la regione V3–V5 si è comportata male nella classificazione delle sequenze appartenenti al phylum Actinobacteria (Fig. 2)., Tendenze simili sono state osservate a livello di genere per i taxa di potenziale rilevanza medica. Sebbene l’intera regione V1-V9 abbia costantemente prodotto i migliori risultati, la regione V6–V9 è stata in particolare la migliore sottoregione per classificare le sequenze appartenenti ai generi Clostridium e Staphylococcus, la regione V3-V5 ha prodotto buoni risultati per Klebsiella e la regione V1-V3 ha prodotto buoni risultati per Escherichia / Shigella (Fig. 2 e Dati di origine).
Infine, la scelta della sub-regione ha influito drammaticamente sul numero di OTU formati durante il clustering di ampliconi in-silico per creare OTU., Quando il clustering a 99% identità di sequenza, tutte le sub-regioni non è riuscito a ricreare il numero di sequenze distinte presenti nel database originale; tuttavia, la regione V4 ancora una volta eseguito peggio (Fig. 1d). In particolare, il numero relativo di OTU prodotte da ciascuna sottoregione non era coerente a soglie di identità diverse(97%, 98%, 99%, Fig.supplementare 3), indicando che il comportamento degli algoritmi di clustering può essere difficile da prevedere quando la quantità di informazioni contenute all’interno di una regione sequenziata è altamente variabile.,
In conclusione, il targeting delle sub-regioni rappresenta un compromesso storico sufficiente per l’identificazione dei taxa a livello di genere o superiore. Tuttavia, il nostro semplice esperimento in-silico dimostra che non è valido presumere che un raggruppamento sempre più fine di queste sub-regioni produrrà la migliore risoluzione tassonomica necessaria per riflettere le specie. Sebbene alcune sottoregioni (ad esempio, V1–V3) forniscano una ragionevole approssimazione della diversità 16S, la maggior parte non cattura variazioni di sequenza sufficienti per discriminare tra taxa strettamente correlati., Notiamo anche che i polimorfismi discriminanti possono essere limitati a specifiche regioni variabili; quindi, alcune sottoregioni saranno più adatte per discriminare membri strettamente correlati di determinati taxa.
Le varianti di copia del gene 16S riflettono la variazione a livello di ceppo
Il clustering di sequenze 16S in OTU ha storicamente servito a due scopi. In primo luogo, ha rimosso varianti di sequenza artefatti minori a causa di amplificazione PCR e sequenziamento errori quando comprimere sequenze in gruppi. In secondo luogo, è crollato varianti di sequenza legittimi che esistono tra taxa batterici strettamente correlati., Anche se quest’ultimo potrebbe non essere sempre desiderabile, è ovvio che non è possibile distinguere tra taxa batterici le cui sequenze 16S variano a una velocità inferiore all’errore riscontrato su una particolare piattaforma di sequenziamento.
Recentemente, i progressi nel CCS hanno notevolmente migliorato i tassi di errore delle piattaforme di sequenziamento a lunga lettura. Allo stesso tempo, i metodi computazionali hanno permesso di distinguere tra variazione di sequenza legittima e artifactual., Questi progressi tecnologici e metodologici significano che i ricercatori hanno ora il potenziale per eseguire un sequenziamento ad alto throughput in grado di rilevare con precisione le varianti a singolo nucleotide sull’intero gene 16S.
Anche se si è tentati di supporre che le varianti a singolo nucleotide possano rappresentare taxa distinti e strettamente correlati, mettiamo in guardia contro questa interpretazione eccessivamente semplicistica dovuta al fatto che molti genomi batterici contengono più copie polimorfiche dei gene12, 13,14 di 16S., Abbiamo eseguito il sequenziamento PacBio CCS di una comunità fittizia batterica di 36 specie (Tabella supplementare 3 e Fig. 4) dimostrare (i) che la sequenza 16S di molti batteri varia tra gli operoni all’interno dello stesso genoma e (ii) che il sequenziamento ad alta velocità è sufficientemente accurato per risolvere queste differenze intragenomiche.
Abbiamo allineato le sequenze PacBio full-length 16S a un database di riferimento contenente una singola sequenza rappresentativa 16S per ogni membro della nostra comunità di simulazione e abbiamo utilizzato le statistiche di allineamento per valutare l’accuratezza di questo approccio di sequenziamento., Confrontando il numero di passaggi utilizzati per generare un CCS con il verificarsi di sostituzioni, inserimenti ed eliminazioni a singolo nucleotide, è stato indicato che dieci passaggi potrebbero ridurre al minimo questi errori combinati ad una frequenza minima di < 1.0% (anche se è stato notevole che l’errore minimo raggiungibile variava tra le esecuzioni di sequenziamento; Fig. 5). Tuttavia, abbiamo osservato una coincidenza di errori di cancellazione con l’omopolimero di posizione eseguito nelle nostre sequenze di riferimento (Fig., 6), che non era specifico per il nucleotide ed era esacerbato dalla lunghezza dell’omopolimero sequenziato (Fig. 7). Successivamente abbiamo convalidato le eliminazioni all’interno del gene Escherichia coli 16S utilizzando il sequenziamento Illumina whole genome shotgun (WGS), che ha dimostrato che solo una delle eliminazioni che si verificano nelle sequenze PacBio era autentica (Fig. 8).,
Soddisfatti del fatto che il sequenziamento CCS possa produrre letture 16S con una bassa frequenza di errori di sostituzione, abbiamo quindi motivato che una percentuale degli errori di sostituzione all’interno di letture accuratamente allineate dovrebbe riflettere la variazione attribuibile ai polimorfismi 16S all’interno del genoma di una specie12. Ad esempio, legge allineato al ceppo di E. coli K-12 substr. MG1655 ha mostrato un profilo di sostituzione, che rispecchiava esattamente quello previsto allineando tutte e sette le sequenze 16S note per essere presenti in questo genome15 (Fig. 2 bis, lettera c)., Siamo stati inoltre in grado di convalidare la stechiometria di queste sostituzioni nucleotidiche quantificando la variazione in letture Illumina WGS allineate in modo comparabile (Fig. 2b) e dimostrare che un profilo di sostituzione simile era riproducibile su più sequenze di sequenziamento (Fig. 9)., Gli allineamenti ad altre sequenze di riferimento nella nostra comunità simulata hanno mostrato una tendenza simile di abbondanti sostituzioni localizzate in specifiche posizioni di base lungo il gene 16S, anche se notiamo che il rapporto segnale-rumore è aumentato significativamente quando il gene 16S in questione aveva meno di 100 letture allineate (Fig. 10).

Polimorfismi nelle sequenze geniche di E. coli 16S rRNA. a La posizione e la frequenza delle sostituzioni che appaiono in E., coli strain K–12 MG1655 V1-V9 amplicons generati dalla nostra comunità finta e sequenziati sulla piattaforma PacBio RS II. b La posizione e la frequenza delle sostituzioni nelle letture generate dal sequenziamento genomico del ceppo isolato di E. coli K-12 MG1655 sulla piattaforma Illumina MiSeq. Le regioni ingrandite mostrano le rispettive posizioni nell’allineamento di tutti i sette geni 16S presenti nel genoma di riferimento di E. coli K-12 MG1655. La sequenza 16S dall’operone rrnD ( * * ) viene utilizzata come riferimento per tutte le fasi SNP. c Il profilo di sostituzione nucleotidica previsto di E., coli K-12 MG1655 basato sull’allineamento delle sette sequenze geniche 16S presenti nel genoma di riferimento. d Il profilo di sostituzione previsto di E. coli O157 Sakai basato sull’allineamento delle sette sequenze geniche 16S presenti nel genoma di riferimento. I pannelli grigi raffigurano regioni variabili definite da siti di legatura del primer comunemente usati (Tabella supplementare 1). Linee tratteggiate indicano la proporzione prevista di sostituzioni nucleotidiche, dato che ci sono sette copie del gene 16S all’interno di ogni genoma., I dati di origine sono forniti come file di dati di origine
L’osservazione che il sequenziamento a lunga lettura può identificare polimorfismi 16S all’interno dello stesso genoma ha importanti implicazioni. In primo luogo,dimostra che non è valido supporre che le letture di sequenze ad alto throughput che differiscono da uno o pochi nucleotidi rappresentino un taxa6, 16 distinto. All’interno di un singolo genoma, due o più sequenze 16S possono essere identiche, mentre altre possono essere uniche., Corrispondentemente, alcuni loci 16S omologhi possono mantenere la sequenza identica tra due ceppi strettamente correlati, mentre altri possono essere divergenti in una o poche posizioni nucleotidiche. In questo contesto, qualsiasi interpretazione a livello comunitario o tassonomico dei dati 16S dovrebbe idealmente tenere conto del fatto che l’abbondanza relativa di sequenze 16S derivanti da taxa strettamente correlati rifletterà una combinazione lineare di (i) la frequenza con cui ogni sequenza unica è rappresentata tra i genomi e (ii) l’abbondanza relativa dei genomi per ciascun taxon.,
In secondo luogo, anche se la variazione di sequenza intragenomica 16S complica l’analisi a livello comunitario, ha anche il potenziale per aumentare la potenza del gene 16S di discriminare tra taxa strettamente correlati, perché consente il confronto basato sulla sequenza di estendersi su più loci divergenti. Ad esempio, esiste una variazione nucleotidica sufficiente per distinguere il ceppo di E. coli K-12 MG1655 dal ceppo enteroemorragico O157 Sakai (Fig. 2 quater, d)., Pertanto, sosteniamo che, se adeguatamente contabilizzate, più copie polimorfiche di 16S non sono un inconveniente da trascurare, piuttosto consentiranno al gene 16S di essere utilizzato nell’analisi del microbioma a livello di ceppo. Notiamo anche che la potenza della variazione di sequenza intragenomica 16S per discriminare taxa strettamente correlati è probabile che diminuisca quando vengono utilizzate sequenze parziali 16S. Ad esempio, SNPS distinguere i ceppi di E. coli K-12 MG1655 (Fig. 2c) da O157 Sakai (Fig. 2d) si trovano nelle regioni variabili V1, V2, V6 e V9.,
I polimorfismi 16S possono essere risolti in vivo
Le comunità di microbiomi sono spesso complesse, esistenti in diversi ambienti biochimici (ad esempio, feci, saliva, espettorato, ecc.) e contenente molte centinaia di taxa unici la cui abbondanza relativa copre un’ampia gamma dinamica. Questa complessità non è ben rappresentata negli esperimenti in-silico o nella comunità simulata. Abbiamo quindi eseguito un ulteriore esperimento per dimostrare che il sequenziamento dell’intero gene 16S mentre si tiene conto degli SNP 16S intragenomici può risolvere taxa batterici strettamente correlati in vivo.,
Abbiamo eseguito il sequenziamento PacBio CCS della regione V1–V9 per quattro campioni di feci umane raccolti da volontari adulti sani. Per confronto, abbiamo sequenziato la regione V1-V3 utilizzando Illumina MiSeq e, per fornire un punto di riferimento per la quantificazione tassonomica a livello di specie, abbiamo eseguito il sequenziamento metagenomico WGS (mWGS) utilizzando Illumina NextSeq. Per valutare la misura in cui ciascuno di questi approcci di sequenziamento può risolvere taxa strettamente correlati, ci siamo concentrati sul genere Bacteroides., Oltre ad essere abbondante nell’intestino umano, questo genere è molto vario e contiene più specie che possono esercitare effetti positivi e negativi sulla salute umana17. È stato anche utilizzato in precedenza come taxon modello per dimostrare l’utilità del gene 16S per l’analisi tassonomica ad alta risoluzione18.
Quando abbiamo calcolato l’abbondanza di Bacteroides a livello di genere, il sequenziamento V1–V9 e il sequenziamento V1–V3 hanno prodotto risultati comparabili., Entrambi gli approcci hanno identificato due individui con bassa abbondanza relativa di Bacteroides (~10-25%) e due individui con alta abbondanza relativa di Bacteroides (~40-60%; Fig. 3 bis). Tuttavia, la quantificazione a livello di specie tramite il sequenziamento mWGS ha rivelato una diversità molto maggiore, con una diversa specie di Bacteroides dominante nell’intestino di ogni individuo (Fig. 3b e Dati integrativi 1). Quando si raggruppano OTU all’identità del 99%, sia il sequenziamento V1–V9 che V1–V3 sono stati in grado di riflettere questa variazione a livello di specie (Fig., 3b), con la notevole eccezione che il sequenziamento V1–V3 non ha rilevato Bacteroides intestinalis, che era abbondante in uno dei quattro campioni di microbioma intestinale umano. Sulla base di questi risultati, concludiamo che, se utilizzati in combinazione con una soglia di identità appropriata (ad esempio, 99%), gli approcci basati su OTU hanno il potenziale per risolvere la diversità a livello di specie osservata nell’intestino umano. Notiamo inoltre che, sebbene il sequenziamento 16S a lunghezza intera possa essere ottimale per l’analisi a livello di specie, anche le regioni variabili altamente informative (ad esempio, V1-V3) possono essere adeguate a questo scopo.,

Rilevamento di Bacteroides in campioni di feci umane. a L’abbondanza relativa del genere Bacteroides in quattro campioni di feci umane quantificati utilizzando ampliconi V1-V9 (asse x) o ampliconi V1–V3 (asse y). b L’abbondanza relativa di specie Bacteroides negli stessi quattro campioni. L’abbondanza di specie è stata quantificata dal sequenziamento mWGS o da OTU V1–V3/V1-V9 generati al 99% di identità., L’abbondanza è indicata per le specie più abbondanti come quantificate da mWGS (per le stime di abbondanza di tutte le specie di Bacteroides rilevate da ciascuna piattaforma, vedere la Tabella supplementare 5). profili di sostituzione del nucleotide c generati allineando tutte le sequenze di amplicone V1-V9 assegnate al singolo OTU identificato come Bacteroides vulgatus. I profili sono mostrati per i due campioni di feci con elevata abbondanza relativa di B. vulgatus (IronHorse e Scott). d Profili di sostituzione nucleotidica previsti dai genomi di riferimento di due diversi ceppi di B. vulgatus ATCC 848239 e mpk40., Sia in c che in d, sono state identificate sostituzioni nucleotidiche rispetto a un singolo gene di riferimento 16S per B. vulgatus ATCC 8482 (gene NCBI ID 5304800). I pannelli grigi raffigurano regioni variabili definite da siti di legatura del primer comunemente usati (Tabella supplementare 1). Linee tratteggiate indicano la proporzione prevista di sostituzioni nucleotidiche, dato che ci sono sette copie del gene 16S all’interno di ogni genoma., Fonte dei dati sono forniti in un file di Dati di Origine
approfittando del fatto che Bacteroides vulgatus era presente ad alta abbondanza relativa in due del nostro microbioma intestinale umano campioni, abbiamo quindi chiesto se intragenomic variazione del gene 16S le copie possono essere rilevati in vivo. Abbiamo allineato ogni sequenza a figura intera classificata come appartenente al nostro B. vulgatus V1–V9 OTUs (Fig. 3b e dati supplementari 1) ad una singola sequenza genetica rappresentativa di B. vulgatus 16S. Abbiamo quindi confrontato i profili di sostituzione dei nucleotidi risultanti (Fig., 3c) con profili previsti da due genomi di riferimento presenti nel database NCBI refseq19 (Fig. 3d).
La maggior parte delle variazioni nucleotidiche presenti nel nostro in vivo ha generato B. vulgatus OTU riflettendo la vera variazione attribuibile ai polimorfismi intragenomici. Al contrario, la variazione probabilmente dovuta a errori di sequenziamento è apparsa bassa e ben al di sotto della frequenza minima di ~14% che ci si aspetterebbe se ci fosse un singolo ceppo B. vulgatus in ciascun campione con sette copie del gene 16S nel suo genoma (Fig. 3c, linee tratteggiate).
Anche se non conoscevamo il vero numero di B., ceppi vulgatus presenti in ogni campione in vivo, è stato notevole che entrambi i profili di sostituzione nucleotidica portavano più somiglianza con ceppo ATCC 8482 rispetto mpk. La variazione esisteva anche in loci specifici che potrebbero potenzialmente indicare differenze significative tra i genomi di riferimento in vivo e ATCC 8482. Ad esempio, un singolo polimorfismo è stato rilevato nella regione V5 di ATCC 8482, che era presente in tre copie 16S (43%). Nel primo campione in vivo (Scott) questo polimorfismo era presente nell ‘ 84% delle letture, mentre nel secondo (IronHorse) era presente nel 69% delle letture., Questi numeri corrispondono strettamente ai numeri attesi se un polimorfismo erano presenti sei e cinque su sette geni 16S, rispettivamente.
In conclusione, mostriamo che il sequenziamento 16S a lunghezza intera del microbioma intestinale umano può risolvere con precisione le sostituzioni a singolo nucleotide che riflettono la variazione intragenomica tra le copie del gene 16S. La presenza di tale variazione indica che le sequenze 16S devono essere raggruppate per riflettere unità tassonomiche significative., Utilizzando OTUs cluster a 99% identità, mostriamo che full-length 16S ha il potenziale per fornire specie e anche la risoluzione tassonomica a livello di ceppo. L’analisi delle comunità microbiche a questi livelli tassonomici promette di fornire una prospettiva molto diversa da quella offerta dalle stime dell’abbondanza a livello di genere.
I polimorfismi intragenomici 16S sono altamente prevalenti
Avendo dimostrato che è possibile risolvere le varianti di copia intragenomiche in vivo, abbiamo quindi cercato di stabilire in che misura tali varianti di copia appaiono nei taxa che si trovano comunemente all’interno del microbioma intestinale umano., Abbiamo inoltre cercato di stabilire se tali profili possono essere utilizzati abitualmente per distinguere tra ceppi della stessa specie.
Abbiamo coltivato 381 taxa dal microbioma intestinale degli individui sani raffigurati in Fig. 3, nonché da altre persone che hanno partecipato allo stesso studio originale20 (Dati supplementari 2). Successivamente abbiamo eseguito il sequenziamento del gene 16S a lunghezza intera su isolati e letture sequenziate allineate per identificare le sostituzioni nucleotidiche caratteristiche delle varianti di copia del gene 16S intragenomiche.,
La classificazione tassonomica degli isolati ha identificato 58 specie putative (Dati supplementari 2), mentre il raggruppamento di una singola sequenza rappresentativa per ciascun isolato con una somiglianza del 99% ha portato a 61 OTU (con tra 1 e 73 isolati assegnati a ciascun OTU). In totale, 349 di 381 isolati sequenziati (54 di 61 OTU) avevano uno o più SNP, indicando la presenza di polimorfismi del gene 16S, e 205 profili SNP unici sono stati identificati quando si tiene conto del potenziale errore di sequenziamento (Fig. 4a e Dati integrativi 2).

Polimorfismi del gene intragenomico 16S negli isolati del microbioma intestinale umano. una posizione di SNPs presente nei geni 16S di isolati batterici coltivati individualmente. Le posizioni SNP sono state identificate attraverso sequenze di geni 16S a lunghezza intera generate per ogni singolo isolato. L’asse X indica la posizione lungo il gene 16S. L’asse Y denota singoli isolati raggruppati in base alla loro filogenesi inferita. La regione blu scuro indica la posizione di un polimorfismo., Per chiarezza, vengono mostrati un massimo di cinque isolati appartenenti alla stessa specie. Per i dettagli sui profili di sostituzione dei nucleotidi per tutti gli isolati sequenziati, vedere Dati supplementari 2. b-d Esempi di profili di sostituzione nucleotidica che mostrano differenze a livello di ceppo tra isolati identificati come appartenenti a tre specie batteriche: b Shigella flexneri; c Bifidobacterium longum; d Collinsella aerofaciens. Per ogni specie, sono mostrati due profili di sostituzione del nucleotide isolato; tuttavia, ulteriori esempi possono essere trovati nei dati supplementari 2., Gli isolati sono stati identificati come appartenenti alla stessa specie se le loro sequenze rappresentative sono state assegnate alla stessa OTU quando si sono raggruppati con un’identità di sequenza del 99%. L’identificazione tassonomica è stata eseguita utilizzando BLAST per allineare sequenze rappresentative al database BLAST NCBI 16S (vedere Metodi). I pannelli grigi raffigurano regioni variabili definite da siti di legatura del primer comunemente usati (Tabella supplementare 1). Linee tratteggiate indicano la proporzione prevista di sostituzioni nucleotidiche, dato il numero di copie del gene 16S previsto per ogni genoma., I dati di origine sono forniti come file di dati di origine
In particolare, il confronto dei profili SNP per gli isolati assegnati allo stesso OTU ha spesso rivelato differenze nella frequenza degli SNP che erano indicative di differenze nelle copie del gene intragenomic 16S tra taxa strettamente correlati. Esempi di diversi profili di sostituzione sono mostrati per tre taxa (Fig. 4b–d), che sono indicativi di variazione del livello di ceppo paragonabile a quello che abbiamo dimostrato in linea di principio per E. coli (Fig. 2 ter).,
In conclusione, mostriamo che molti dei membri culturabili del microbioma intestinale umano possiedono spesso polimorfismi del gene 16S, che, se correttamente contabilizzati, hanno il potenziale per risolvere ceppi della stessa specie.