het volledige 16S gen biedt een betere taxonomische resolutie
Het ~1500 bp 16S rRNA gen bestaat uit negen variabele gebieden afgewisseld door de sterk geconserveerde 16S sequentie (Fig. 1 bis). Het rangschikken van het volledige gen werd oorspronkelijk bereikt door het rangschikken van Sanger., Dit vereiste het klonen genen, het produceren, en het assembleren van twee aan drie leest per kloon, en het veroorzaken van beperkte bemonsteringsdiepte bij hoge kosten en inspanning. Momenteel, echter, de overgrote meerderheid van de onderzoeksopeenvolging slechts een deel van het gen, omdat het wijd gebruikte Illumina rangschikkende platform (hogere productie, lagere kosten, verminderde inspanning vergeleken met Sanger) korte opeenvolgingen ( ≤ 300 basissen) veroorzaakt., De verschillende subgebieden van het gen worden daarom gericht, die zich van enige veranderlijke gebieden, zoals V4 of V6, aan drie veranderlijke gebieden, zoals V1–V3 of V3–V5 (in het menselijke Microbiome Project samen met het 454 rangschikken platform9) uitstrekken.
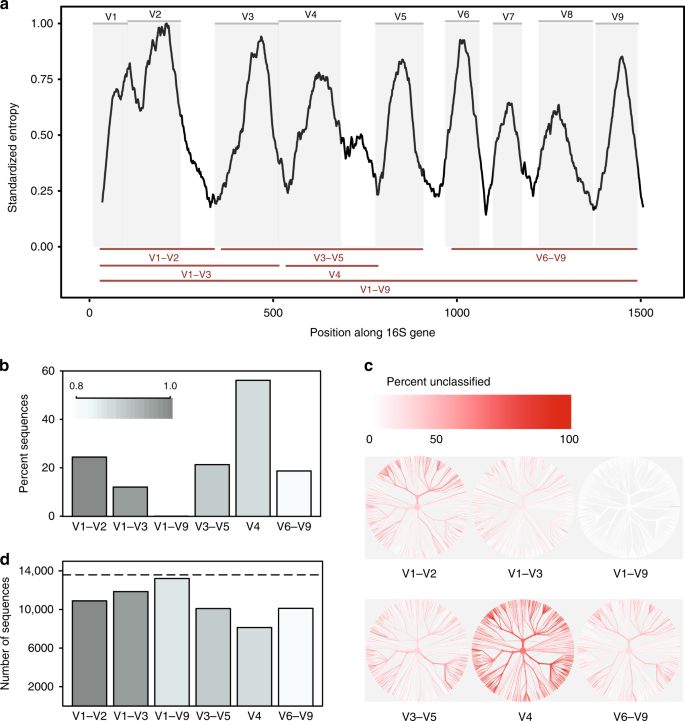
In-silico vergelijking van 16S rRNA variabele regio ‘ s. een Shannon entropie over het 16S gen gebaseerd op de uitlijning van een enkele representatieve sequentie voor elke bekende species aanwezig in de Greengenes database., Sequenties werden afgestemd op één enkel referentie 16S gen voor Escherichia coli K-12 MG1655 (NCBI Gen ID 947777). Grijze panelen tonen variabele gebieden gedefinieerd door veelgebruikte primer-bindingsplaatsen (aanvullende tabel 1). Variabele regio ‘ s die in deze studie worden beschouwd worden weergegeven als rode lijnen (onder). b percentage sequenties voor elk variabel gebied dat niet kon worden geïdentificeerd tot soortenniveau bij het classificeren van elke sequentie aan de hand van de referentiedatabank waaruit deze is afgeleid bij een betrouwbaarheidsdrempel van 80% (RDP-classifier)., C bomen gebaseerd op taxonomie van sequenties aanwezig in de in-silico database. Dezelfde boom wordt verstrekt voor elke variabele regio. De kleur van elke tak weerspiegelt het aandeel van sequenties binnen elke clade die niet konden worden geïdentificeerd aan soort niveau. d het aantal OTUs dat wordt gecreeerd wanneer het clusteren opeenvolgingen voor elk veranderlijk gebied bij 99% opeenvolgingsgelijkenis. Stippellijn geeft het aantal unieke sequenties aan (>1% verschillend) in de oorspronkelijke database., Brongegevens worden geleverd als een brongegevensbestand
wij stellen dat het targeting van subregio ‘ s een historisch compromis is, als gevolg van technologiebeperkingen.10 Vandaag, zijn zowel PacBio als Oxford Nanopore het rangschikken platforms geschikt om routinematig leest boven 1500 bp te produceren en wordt het hoog-productie rangschikken van het volledige 16S gen steeds overwegend., Daarom stellen wij voor dat de rechtvaardiging voor dit compromis moet worden herzien en hebben wij een eenvoudig in-silico-experiment uitgevoerd om het voordeel van volledige 16S-sequencing ten opzichte van het targeting van subregio ‘ s aan te tonen.
we hebben een set niet-redundante (d.w.z., > 1% verschillend), volledige 16S-sequenties gedownload van een openbare database (Greengenes)., Voordeel nemend van het feit dat een substantieel deel van deze opeenvolgingen PCR-primer-bindende plaatsen opnam, trimde wij hen om in-silico amplicons voor verschillende subgebieden te produceren, op de plaats van PCR-inleidingen die algemeen in microbiome studies worden gebruikt (Fig. 1a en aanvullende tabellen 1-2)., Aangenomen dat elke sequentie in onze gedownloade database een unieke soort vertegenwoordigde, gebruikten we vervolgens een gemeenschappelijke classificatiebenadering (het ribosoom Database Project (RDP) classifier11) om de frequentie te berekenen waarmee in-silico amplicons voor elke subregio accurate taxonomische classificatie op soortniveau konden bieden (met behulp van de originele database als referentie). In een tweede experiment hebben we ook onze in-silico amplicons geclusterd om OTUs te genereren bij verschillende, veelgebruikte, sequentievergelijkingsdrempels(97%, 98%, 99%).,
We vonden dat subregio ‘ s aanzienlijk verschilden in de mate waarin zij vol vertrouwen onderscheid konden maken tussen de volledige 16S-sequenties die worden gebruikt om soorten voor te stellen (Fig. 1 ter). De V4-regio presteerde het slechtst: 56% van de in-silico-amplicons voldeed niet met vertrouwen aan de volgorde van oorsprong op dit taxonomische niveau. Wanneer daarentegen een volledige lengte-sequentie met alle variabele gebieden werd gebruikt, was het mogelijk om bijna alle sequenties als de juiste soort te classificeren (aanvullende Fig. 1 bis)., De wijziging van databases en classificatiedrempels beïnvloedden het percentage in-silico amplicons dat nauwkeurig kon worden afgestemd, maar beïnvloedden de heersende trends niet (Aanvullende Fig. 1 bis, b).
ten tweede vertoonden verschillende subregio ‘ s bias in de bacteriële taxa die zij konden identificeren (Fig. 1c). Bijvoorbeeld, voerde het gebied V1-V2 slecht uit bij het classificeren van opeenvolgingen die tot de phylum Proteobacteria behoren, terwijl het gebied V3-V5 slecht bij het classificeren van opeenvolgingen die tot de phylum Actinobacteria behoren (aanvullende Fig. 2)., Vergelijkbare trends werden gezien op geslachtsniveau voor taxa die mogelijk medisch relevant zijn. Hoewel de volledige Regio V1-V9 consequent de beste resultaten opleverde, was de regio V6–V9 met name de beste subregio voor het classificeren van sequenties behorend tot de geslachten Clostridium en Staphylococcus, leverde de regio V3–V5 goede resultaten op voor Klebsiella, en de regio V1–V3 leverde goede resultaten op voor Escherichia/Shigella (aanvullende Fig. 2 en brongegevens).
ten slotte had de keuze van de subregio een dramatische invloed op het aantal OTUs dat gevormd werd bij het clusteren van In-silico amplicons om OTUs te creëren., Bij het clusteren op 99% sequentieidentiteit, slaagden alle subregio ‘ s er niet in om het aantal verschillende sequenties in de oorspronkelijke database opnieuw te maken; echter, de V4-regio presteerde opnieuw het slechtst (Fig. 1d). Met name was het relatieve aantal Otu ‘ s dat door elke subregio werd geproduceerd, niet consistent bij verschillende identiteitsdrempels (97%, 98%, 99%, aanvullende Fig. 3), die aangeeft dat het gedrag van clustering algoritmen moeilijk te voorspellen kan zijn wanneer de hoeveelheid informatie binnen een sequenced gebied zeer variabel is.,
concluderend kan worden gesteld dat het targeten van subregio ‘ s een historisch compromis is dat voldoende was voor de identificatie van taxa op geslachtsniveau of hoger. Ons eenvoudige In-silico-experiment toont echter aan dat het niet geldig is om aan te nemen dat een steeds fijnere clustering van deze subregio ‘ s zal resulteren in de verbeterde taxonomische resolutie die nodig is om soorten weer te geven. Hoewel sommige subregio ’s (bijvoorbeeld V1–V3) een redelijke benadering van de diversiteit van 16′ s bieden, vangen de meeste niet voldoende sequentievariaties om onderscheid te maken tussen nauw verwante taxa., We merken ook op dat discriminerende polymorfismen beperkt kunnen blijven tot specifieke variabele regio ‘s; zo zullen bepaalde subregio’ s beter geschikt zijn voor het discrimineren van nauw verwante leden van bepaalde taxa.
16S Gene copy varianten weerspiegelen stam-niveau variatie
Clustering van 16S sequenties in OTUs heeft historisch twee doeleinden gediend. Ten eerste heeft het kleine artifactual opeenvolgingsvarianten verwijderd als gevolg van PCR-versterking en het rangschikken van fouten bij het samenvouwen van opeenvolgingen in groepen. Ten tweede, het is ingestort legitieme sequentievarianten die bestaan tussen nauw verwante bacteriële taxa., Hoewel de laatste misschien niet altijd wenselijk zijn, is het logisch dat u geen onderscheid kunt maken tussen bacteriële taxa waarvan de 16S-sequenties variëren met een tarief dat lager is dan de fout die op een bepaald sequencingplatform wordt aangetroffen.
recente ontwikkelingen in CCS hebben de foutenpercentages van langlezen sequencingplatforms drastisch verbeterd. Tegelijkertijd, computationele methoden hebben het mogelijk gemaakt om onderscheid te maken tussen legitieme Versus artifactuele sequentie variatie., Deze technologische en methodologische vooruitgang betekenen onderzoekers hebben nu het potentieel om hoog-productie het rangschikken uit te voeren dat single-nucleotidevarianten over het volledige gen van 16S nauwkeurig kan ontdekken.
hoewel het verleidelijk is om aan te nemen dat een-nucleotide varianten verschillende, nauw verwante taxa kunnen vertegenwoordigen, waarschuwen we tegen deze al te simplistische interpretatie vanwege het feit dat veel bacteriële genomen meerdere polymorfe kopieën van de 16S gene12,13,14 bevatten., We voerden Pacb CCS sequencing van een 36 soorten bacteriële mock gemeenschap (aanvullende tabel 3 en aanvullende Fig. 4) om aan te tonen (i) dat de 16S-opeenvolging van vele bacteriën tussen operonen binnen hetzelfde genoom varieert en (ii) dat het rangschikken van de hoog-productie voldoende nauwkeurig is om deze intragenomic verschillen op te lossen.
We hebben PacBio full-length 16S-sequenties afgestemd op een referentiedatabase met een enkele representatieve 16S-sequentie voor elk lid van onze mock-gemeenschap en hebben de alignmentstatistieken gebruikt om de nauwkeurigheid van deze sequencingbenadering te evalueren., Het vergelijken van het aantal passages gebruikt om een CCS te genereren met het voorkomen van single-nucleotide substituties, inserties en deleties gaf aan dat tien passes deze gecombineerde fouten konden minimaliseren tot een minimale frequentie van < 1,0% (hoewel het opmerkelijk was dat de minimaal haalbare fout varieerde tussen sequencing runs; aanvullende Fig. 5). We hebben echter wel een samenloop van verwijderingsfouten waargenomen met de locatie homopolymeer loopt in onze referentie sequenties (aanvullende Fig., 6), die niet nucleotide-specifiek was en werd verergerd door de lengte van het gesequenced homopolymeer (aanvullende Fig. 7). Vervolgens valideerden we deleties binnen het Escherichia coli 16S gen met behulp van Illumina whole genome shotgun (WGS) sequencing, wat aantoonde dat slechts één van de deleties die voorkomen in PacBio sequenties echt was (aanvullende Fig. 8).,tevreden dat CCS-sequencing 16S-reads kan produceren met een lage frequentie van substitutiefouten, redeneerden we vervolgens dat een deel van de substitutiefouten binnen nauwkeurig uitgelijnde reads variatie moet weerspiegelen die kan worden toegeschreven aan 16S-polymorfismen binnen een soortengenome12. Bijvoorbeeld, leest uitgelijnd op de E. coli stam K-12 substr. MG1655 toonde een substitutieprofiel, dat precies het voorspelde weerspiegelde door alle zeven van de 16S-sequenties waarvan bekend is dat ze aanwezig zijn in dit genoom15 uit te lijnen (Fig. 2a, c)., We waren verder in staat om de stoichiometrie van deze nucleotide substituties te valideren door variatie in vergelijkbaar uitgelijnde Illumina WGS reads te kwantificeren (Fig. 2b) en aantonen dat een vergelijkbaar substitutieprofiel reproduceerbaar was over meerdere sequencing runs (aanvullende Fig. 9)., Alignments naar andere referentie sequenties in onze mock gemeenschap toonde een soortgelijke trend van overvloedige substituties gelokaliseerd op specifieke basisposities langs het 16S gen, hoewel we merken dat de signaal-ruisverhouding aanzienlijk toenam het 16S gen in kwestie had minder dan 100 uitgelijnde reads (aanvullende Fig. 10).

polymorfismen in E. coli 16S rRNA gensequenties. a de positie en frequentie van substituties die voorkomen in E., coli stam K-12 MG1655 V1-V9 amplicons gegenereerd uit onze mock community en gesequenced op het PacBio RS II platform. b de positie en frequentie van substituties in reads gegenereerd door genomische sequencing van de geïsoleerde E. coli stam K-12 MG1655 op het Illumina MiSeq platform. De vergrote gebieden tonen respectieve posities in de uitlijning van alle zeven 16S genen huidig in het E. coli K-12 MG1655 verwijzingsgenoom. De 16S-sequentie van het rrnd operon ( * * ) wordt gebruikt als referentie voor alle SNP-fasering. c het voorspelde nucleotide substitutieprofiel van E., coli K-12 MG1655 gebaseerd op het uitlijnen van de zeven 16S genopeenvolgingen huidig in het verwijzingsgenoom. d het voorspelde substitutieprofiel van E. coli O157 Sakai gebaseerd op het uitlijnen van de zeven 16S gensequenties aanwezig in het referentiegenoom. Grijze panelen tonen variabele gebieden gedefinieerd door veelgebruikte primer-bindingsplaatsen (aanvullende tabel 1). De gestippelde lijnen wijzen op het verwachte aandeel van nucleotide substituties, gegeven zijn er zeven 16S genkopieën binnen elk genoom., Brongegevens worden verstrekt als een brongegevensbestand
de observatie dat lange-readsequencing 16S-polymorfismen binnen hetzelfde genoom kan identificeren, heeft belangrijke implicaties. Ten eerste,het toont aan dat het niet geldig is om aan te nemen dat high-throughput sequentie verschillend door één of enkele nucleotiden een verschillende taxa6, 16 vertegenwoordigt. Binnen één enkel genoom, kunnen twee of meer 16S opeenvolgingen identiek zijn, terwijl anderen uniek kunnen zijn., Dienovereenkomstig, kunnen sommige homologe 16S loci identieke opeenvolging tussen twee nauw verwante spanningen behouden, terwijl anderen bij één of weinig nucleotideposities kunnen hebben gedivergeerd. In dit verband moet elke taxonomische interpretatie op communautair niveau van 16S-gegevens idealiter rekening houden met het feit dat de relatieve abundantie van 16S-sequenties die voortvloeit uit zeer nauw verwante taxa een lineaire combinatie zal weerspiegelen van i) de frequentie waarmee elke unieke sequentie wordt weergegeven over genomen en ii) de relatieve abundantie van de genomen voor elk taxon.,
ten tweede, hoewel intragenomic 16S sequentievariatie de analyse op Gemeenschapsniveau compliceert, heeft het ook het potentieel om het vermogen van het 16S gen te vergroten om onderscheid te maken tussen nauw verwante taxa, omdat het sequentiegebaseerde vergelijking mogelijk maakt om zich uit te breiden over meerdere uiteenlopende loci. Er bestaat bijvoorbeeld voldoende nucleotidevariatie om E. coli stam K-12 MG1655 te onderscheiden van de enterohemorragische stam O157 Sakai (Fig. 2c, d)., Aldus, stellen wij dat, wanneer goed verantwoord, veelvoudige polymorfe 16S exemplaren geen ongemak zijn om over het hoofd te worden gezien, eerder zullen zij het 16S gen toelaten om in de microbiome analyse van het STAM-niveau worden gebruikt. Wij merken ook op dat de macht van intragenomic 16S opeenvolgingsvariatie om nauw verwante taxa te onderscheiden waarschijnlijk zal verminderen wanneer gedeeltelijke 16S opeenvolgingen worden gebruikt. Bijvoorbeeld, SNP ‘ s onderscheidend de E. coli stammen K-12 MG1655 (Fig. 2c) van O157 Sakai (vijg. 2d) worden gevonden in variabele regio ‘ s V1, V2, V6 en V9.,
16S polymorfismen kunnen in vivo worden opgelost
Microbioomgemeenschappen zijn vaak complex en bestaan in diverse biochemische omgevingen (bijv. ontlasting, speeksel, sputum, enz.) en met vele honderden unieke taxa waarvan de relatieve abundantie een breed dynamisch bereik overspant. Deze complexiteit is niet goed vertegenwoordigd in zowel in-silico als nep-communautaire experimenten. Daarom voerden we een extra experiment uit om aan te tonen dat het rangschikken van het volledige 16S-gen terwijl de boekhouding voor intragenomic 16s SNPs nauw verwante bacteriële taxa in vivo kan oplossen.,
We hebben Paccio CCS sequencing van de V1–V9 regio uitgevoerd voor vier menselijke ontlasting monsters verzameld bij gezonde volwassen vrijwilligers. Ter vergelijking hebben we de V1–V3 regio gesequenced met behulp van de Illumina MiSeq en, om een benchmark te bieden voor taxonomische kwantificering op soortenniveau, hebben we metagenomic WGS (mWGS) sequencing uitgevoerd met behulp van de Illumina NextSeq. Om de mate te evalueren waarin elk van deze sequencing benaderingen nauw verwante taxa kan oplossen, richtten we ons op het geslacht Bacteroides., Naast het feit dat overvloedig in de menselijke darm, dit geslacht is zeer divers, met meerdere soorten die zowel goede als slechte effecten op de menselijke gezondheid kan uit te oefenen 17. Het is ook eerder gebruikt als model taxon voor het aantonen van het nut van het 16S-gen voor taxonomische analyse met hoge resolutie 18.
toen we Bacteroides abundantie berekenden op genus niveau, produceerden v1–V9 sequencing en V1-V3 sequencing vergelijkbare resultaten., Beide benaderingen identificeerden twee individuen met lage Bacteroides relatieve overvloed (~10-25%) en twee individuen met hoge Bacteroides relatieve overvloed (~40-60%; Fig. 3a). Nochtans, onthulde de species-niveau kwantificering via mWGS het rangschikken veel grotere diversiteit, met een verschillende Bacteroides species dominant in de darm van elk individu (Fig. 3b en aanvullende gegevens 1). Bij het clusteren van OTUs bij 99% – identiteit, waren zowel v1–V9 als v1–V3 sequencing in staat om deze variatie op soortenniveau weer te geven (Fig., 3b), met de opmerkelijke uitzondering dat V1–V3 het rangschikken Bacteroides intestinalis niet ontdekte, die overvloedig in één van de vier menselijke steekproeven van darmmicrobioom was. Op basis van deze resultaten concluderen we dat, wanneer gebruikt in combinatie met een passende identiteitsdrempel (bijvoorbeeld 99%), OTU-gebaseerde benaderingen het potentieel hebben om soortendiversiteit waargenomen in de menselijke darm op te lossen. We merken verder op dat, hoewel de volledige lengte 16S sequencing optimaal kan zijn voor species-level analyse, zeer informatieve variabele gebieden (bijv., V1-V3) ook geschikt voor dit doel kunnen zijn.,

detecteren van Bacteroides in menselijke ontlastingsmonsters. a de relatieve abundantie van het geslacht Bacteroides in vier menselijke ontlastingsmonsters gekwantificeerd met behulp van V1–V9 amplicons (x-as) of V1–V3 amplicons (y-as). b De relatieve abundantie van Bacteroides-soorten in dezelfde vier monsters. De dichtheid van de Species werd gekwantificeerd door het rangschikken van mWGS of Van V1–V3/V1–V9 OTUs die bij 99% – identiteit wordt gegenereerd., Abundantie wordt getoond voor de meest voorkomende soorten zoals gekwantificeerd door MWG ‘ s (voor abundantieschattingen van alle Bacteroides-soorten die door elk platform worden gedetecteerd, zie aanvullende tabel 5). C Nucleotide substitutie profielen gegenereerd door het uitlijnen van alle v1-V9 Amplicon sequenties toegewezen aan de enige OTU geïdentificeerd als Bacteroides vulgatus. Profielen worden getoond voor de twee ontlasting monsters met een hoge B. vulgatus relatieve abundantie (IronHorse en Scott). d Nucleotide substitutieprofielen voorspeld op basis van de referentie genomen van twee verschillende B. vulgatus stammen ATCC 848239 en mpk40., In zowel c als d werden nucleotidesubstituties geïdentificeerd ten opzichte van een enkel referentie-16S-gen voor B. vulgatus ATCC 8482 (NCBI-Gen ID 5304800). Grijze panelen tonen variabele gebieden gedefinieerd door veelgebruikte primer-bindingsplaatsen (aanvullende tabel 1). De gestippelde lijnen wijzen op het verwachte aandeel van nucleotide substituties, gegeven zijn er zeven 16S genkopieën binnen elk genoom., Brongegevens worden geleverd als een brongegevensbestand
gebruikmakend van het feit dat Bacteroides vulgatus aanwezig was bij een hoge relatieve abundantie in twee van onze menselijke darmmicrobioommonsters, vroegen we vervolgens of intragenomische variatie tussen 16S-genkopieën in vivo kon worden gedetecteerd. We hebben elke volledige lengte sequentie ingedeeld als behorend tot onze B. vulgatus V1-V9 OTUs (Fig. 3b en aanvullende gegevens 1) tot één enkele representatieve B. vulgatus 16S-gensequentie. Vervolgens vergeleken we de resulterende nucleotide substitutieprofielen (Fig., 3c) met voorspelde profielen op basis van twee referentie genomen aanwezig in de NCBI RefSeq database19 (Fig. 3d).
de meerderheid van de nucleotide variatie aanwezig in onze in vivo gegenereerde B. vulgatus OTU weerspiegelde echte variatie toe te schrijven aan intragenomische polymorfismen. In tegenstelling, variatie waarschijnlijk te wijten aan sequencing fouten bleek laag en ver onder de minimale ~14% frequentie die zou worden verwacht als er een enkele B. vulgatus stam in elk monster met zeven 16S gen kopieën in zijn genoom (Fig. 3c, gestippelde lijnen).
hoewel we het ware getal van B. niet kenden., vulgatus stammen aanwezig in elk in-vivo monster, was het opmerkelijk dat beide nucleotide substitutie profielen meer gelijkenis vertonen met stam ATCC 8482 dan mpk. De variatie bestond ook bij specifieke loci die potentieel betekenisvolle verschillen tussen de in vivo en ATCC 8482 verwijzingsgenomen kon aangeven. Bijvoorbeeld, werd één enkel polymorfisme ontdekt in het gebied V5 van ATCC 8482, dat in drie 16S exemplaren (43%) aanwezig was. In het eerste in-vivo Monster (Scott) was dit polymorfisme aanwezig in 84% van de reads, terwijl in het tweede (IronHorse) het aanwezig was in 69% van reads., Deze aantallen corresponderen nauw met de aantallen die worden verwacht als een polymorfisme aanwezig zes en vijf van zeven 16S genen, respectievelijk waren.
concluderend tonen we aan dat volledige 16S-sequencing van het menselijke darmmicrobioom nauwkeurig single-nucleotide substituties kan oplossen die intragenomische variatie tussen 16S-genkopieën weerspiegelen. De aanwezigheid van dergelijke variatie wijst erop dat 16S-sequenties moeten worden geclusterd om zinvolle taxonomische eenheden weer te geven., Met OTUs geclusterd op 99% identiteit, laten we zien dat full-length 16 ‘ s heeft het potentieel om soorten en zelfs stam-niveau taxonomische resolutie. Analyse van microbiële gemeenschappen op deze taxonomische niveaus belooft een heel ander perspectief te bieden dan het perspectief dat wordt geboden door genus-niveau abundantieschattingen.
Intragenomische 16 ‘ s polymorfismen zijn zeer overwegend
nadat is aangetoond dat het mogelijk is om intragenomische kopieervarianten in vivo op te lossen, hebben we vervolgens getracht vast te stellen in welke mate dergelijke kopieervarianten voorkomen in taxa die gewoonlijk worden aangetroffen in het menselijke darmmicrobioom., Verder hebben we getracht vast te stellen of dergelijke profielen routinematig kunnen worden gebruikt om stammen van dezelfde soort te onderscheiden.
we kweekten 381 taxa uit het darmmicrobioom van de gezonde individuen afgebeeld in Fig. 3, alsmede van andere personen die aan dezelfde oorspronkelijke studie deelnemen20 (aanvullende gegevens 2). Vervolgens voerden we het volledige 16S gensequencing uit op isolaten en uitgelijnde sequenced reads om nucleotide substituties te identificeren die kenmerkend zijn voor intragenomic 16S genexemplaarvarianten.,
taxonomische classificatie van isolaten identificeerde 58 veronderstelde soorten (aanvullende gegevens 2), terwijl het clusteren van een enkele representatieve sequentie voor elk isolaat met een gelijkenis van 99% resulteerde in 61 OTUs (met tussen 1 en 73 isolaten toegewezen aan elke OTU). In totaal hadden 349 van 381 gesequenced isolaten (54 Van 61 OTUs) een of meer SNP, wat wijst op de aanwezigheid van 16S genpolymorfismen, en 205 unieke SNP-profielen werden geïdentificeerd bij het in aanmerking nemen van mogelijke sequentiefouten (Fig. 4a en aanvullende gegevens 2).

Intragenomische 16S genpolymorfismen in humane darmmicrobioomisolaten. een locatie van SNPs aanwezig in de 16S genen van individueel gekweekte bacteriële isolaten. SNP-locaties werden geà dentificeerd door phasing full-length 16S gensequenties gegenereerd voor elk individueel isolaat. De x-as Geeft positie langs het 16S-gen aan. Y-as geeft individuele isolaten geclusterd op basis van hun afgeleide fylogenie. Donkerblauw gebied geeft de locatie van een polymorfisme aan., Voor de duidelijkheid worden maximaal vijf isolaten van dezelfde soort getoond. Zie aanvullende gegevens 2 voor details van nucleotidesubstitutieprofielen voor alle gesequenced isolaten. b-d voorbeelden van nucleotide substitutieprofielen die verschillen in stamniveau vertonen tussen isolaten die tot drie bacteriesoorten behoren: B Shigella flexneri; c Bifidobacterium longum; d Collinsella aerofaciens. Voor elke soort worden twee isolaat-nucleotide-substitutieprofielen getoond; aanvullende voorbeelden zijn echter te vinden in aanvullende gegevens 2., Isolaten werden geïdentificeerd als behorend tot dezelfde soort als hun representatieve sequenties werden toegewezen aan dezelfde OTU bij het clusteren op 99% sequentie-identiteit. Taxonomische identificatie werd uitgevoerd met behulp van BLAST om representatieve sequenties af te stemmen op de NCBI 16S BLAST database (zie methoden). Grijze panelen tonen variabele gebieden gedefinieerd door veelgebruikte primer-bindingsplaatsen (aanvullende tabel 1). Gestippelde lijnen geven het verwachte aandeel van nucleotide substituties, gezien het aantal 16S gen kopieën voorspeld voor elk genoom., Brongegevens worden verstrekt als een Brongegevenbestand
met name bij het vergelijken van SNP-profielen voor isolaten die aan dezelfde OTU zijn toegewezen, werden vaak verschillen in de frequentie van SNP ‘ s gevonden die wijzen op verschillen in intragenomische 16S-genkopieën tussen nauw verwante taxa. Voorbeelden van verschillende substitutieprofielen worden getoond voor drie taxa (Fig. 4b-d), die wijzen op variatie op stamniveau, vergelijkbaar met wat we in principe voor E. coli hebben aangetoond (Fig. 2b).,tot slot tonen we aan dat veel van de cultureerbare leden van het menselijke darmmicrobioom vaak 16S-genpolymorfismen bezitten, die, wanneer goed verantwoord, het potentieel hebben om stammen van dezelfde soort op te lossen.