RNA-sekvensering (Wang 2009) ersätter snabbt genuttryck mikroarrays i många laboratorier. RNA-seq kan du kvantifiera, upptäcka och profil rna. För denna teknik omvandlas mRNA (och andra RNA) först till cDNA. CDNA används sedan som ingång för en nästa generations sekvenseringsbibliotek förberedelse. I den här artikeln ska jag ge en kort genomgång av RNA-seq och introducera de viktigaste metoderna som används idag.
Varför Är RNA-Seq ”Bättre” Än Microarrays?,
det finns flera fördelar RNA-seq har över mikroarrays:
med RNA-seq kan du förhöra mer än bara differentiellt genuttryck. Även om det finns mikroarrays tillgängliga för exon-level och microRNA analys, de flesta användare är fortfarande intresserade av grundläggande, förmodligen 3 ’ partisk, differentialgen uttryck. Med RNA-seq kan du titta på kodning och icke-kodande RNA, vid skarvning och allel specifikt uttryck, och eventuellt snart vid fullängds cDNA-sekvenser, vilket eliminerar behovet av att härleda eller montera isoformer.,
Microarrays är också partiska, eftersom vi måste bestämma vilket innehåll som ska placeras på matrisen. Eftersom RNA-seq inte använder sonder eller primrar, lider data av mycket lägre fördomar (även om jag inte menar att RNA-seq inte har någon).
RNA-seq tillhandahåller digitala data i form av anpassade läsantal, vilket resulterar i ett mycket brett dynamiskt område, vilket förbättrar detektionens känslighet för sällsynta transkript.
det är också mycket kostnadseffektivt att microarrays, som idag, mellan 6-30 prover kan multiplexeras i en enda Illumina sekvensering körfält.,
slutligen kan du analysera ett RNA-seq-dataset igen eftersom mer information om transkriptomen blir tillgänglig. Om ett papper publiceras som visar en intressant splice-variant i ett liknande system som det du arbetar med, kanske du vill gå tillbaka och titta på den splitsningen i dina prover; och du skulle redan ha data att göra det.
hur verkar RNA-Seq?
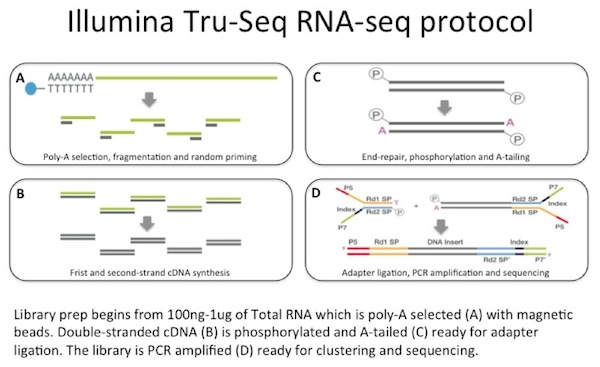
det finns många metoder för att utföra ett RNA-seq-experiment. Faktum är att teknikerna utvecklas så snabbt att det kan vara svårt att bestämma vilken som ska användas., Ett grundläggande val är mellan 1) Random-primed cDNA-syntes från dubbelsträngade cDNA eller 2) rna-ligationsmetoder (granskade och jämförda i Levin 2010). De flesta använder den första metoden och måste sedan göra ett ytterligare val mellan ett strängspecifikt protokoll och ett som inte är. Den metod som används mest i mitt labb är Illuminas TruSeq RNA-seq, som är ett slumpmässigt grundat cDNA-syntes icke-strängspecifikt protokoll.
När du har ett sekvenseringsbibliotek är det sekvenserat till ett angivet djup, vilket är beroende av vad du vill göra med data., Dessa läsningar är inriktade på genomet eller transkriptomet och räknas för att bestämma differentialgenuttryck eller ytterligare analyseras för att bestämma splitsning och isoformuttryck. De flesta människor sekvenserar RNA med parade-end 50-100bp metoder. Undantaget är micrna-sekvensering, eftersom detta endast kräver enstaka 36bp-sekvensering i de flesta fall.
vår RNA-Seq-metod
vi använder mellan 100 ng till 1 µg total RNA som ingång till en mRNA-infångning med oligo-dT belagda magnetiska pärlor. MRNA är fragmenterad, och sedan utförs en slumpmässig primerad cDNA-syntes., Den resulterande dubbel-strand cDNA används som ingång till en standard Illumina bibliotek prep som inkluderar end-reparation, adapter ligering och PCR-amplifiering för att ge dig ett bibliotek redo för sekvensering.
varför bry sig om Strand Information?
det har diskuterats mycket om anti-sense transkription och dess biologiska relevans. Om du är intresserad av enkla differentialgen uttryck, kommer strand information inte lägga mycket till ditt experiment, men kommer att göra ditt protokoll mer komplex., Med detta sagt kan du utföra den mest antagna metoden utan för mycket extra ansträngning. För att göra detta, under 2: a sträng cDNA-syntes, använd uracil för inkorporering istället för tymin. Följ Illumina library prep som vanligt, men efter adapterligering och före PCR-förstärkning tillsätt uracil-DNA glykosylas för att försämra 2ndstrand. Detta resulterar i alla läser börjar i samma riktning så att du kan avgöra vilken sträng som transkriberas i ditt prov.
Vad Kan Du Egentligen Göra Med RNA-Seq?,
RNA-seq är ett kraftfullt och mångsidigt verktyg som har publicerats i stor utsträckning under de senaste åren. Jag har valt ett par av mina favoriter (några från arbete som utförs i kärnanläggningen jag hanterar) för att illustrera vad du kan göra med RNA-sekvensering.
- Jabbari, et al. används RNA-seq för att undersöka psoriasis och hitta nya gener för funktionell analys. De jämförde sina RNA-seq-data med publicerade matrisstudier och hittade 1700 nya kandidater. Dessa validerades av qPCR, och jämförelse med funktionella databaser för psoriasis stödde deras roll i patogenesen.
- kutter, et al., används RNA-seq i en studie som undersöker bevarandet av RNA-polymeras III-bindning hos däggdjur för att validera uttryck av gener som upptas av Pol III som analyseras av ChIP-seq.
- Mercer et al. kombinerad RNA-seq och mikroarray-baserad fångst för att identifiera och karakterisera sällsynta transkript, som normalt är odetekterbara. De riktade transkripten ökade i sekvens läs överflöd från 0.21% Pre-capture till 80% post capture., De fann mer än 200 tidigare oannoterade isoformer för nästan 50 proteinkodande loci, inklusive ett nytt alternativ isoform av TP53, vilket är en mycket väl karakteriserad gen. Detta tyder på att det fortfarande finns mycket komplexitet i genomet och transkriptomet som ska lösas.
Sammanfattningsvis är RNA-seq fortfarande ett verktyg som utvecklas, men är i de flesta fall att föredra framför mikroarrayer. Det är känsligare, mer robust och kan vara mer kostnadseffektivt. Vilka RNA-seq-projekt planerar du nu för ditt projekt?,
Jabbari et al: transkriptionell profilering av Psoriasis med användning av RNA-seq avslöjar tidigare oidentifierade differentiellt uttryckta gener. Journal of Investigative Dermatology 2011.
kutter et al: Pol III bindning i sex däggdjur visar bevarande bland aminosyraisotyper trots divergens mellan tRNA gener. Nature Genetics 2011.
Levin et al: omfattande jämförande analys av strandspecifika rna-sekvenseringsmetoder. NaturMetoder 2010.
Mercer et al: målinriktad RNA-sekvensering avslöjar den mänskliga transkriptomens djupa komplexitet. Nature Biotechnology 2012.,
Wang et al: RNA-Seq: ett revolutionerande verktyg för transcriptomics. Nat Rev Genet 2009.
publicerades ursprungligen den 25 maj 2012. Uppdaterad och reviderad den 16 augusti 2015.

har detta hjälpt dig? Vänligen dela med ditt nätverk.