den fullständiga 16S-genen ger bättre taxonomisk upplösning
~1500 bp 16S rRNA-genen består av nio variabla regioner som varvas genom den högt konserverade 16S-sekvensen (Fig. 1a). Sekvensering av hela genen uppnåddes ursprungligen av Sanger-sekvensering., Detta krävde kloninggener, generering och montering av två till tre läser per klon och producerar begränsat provtagningsdjup till hög kostnad och ansträngning. För närvarande, emellertid, den stora majoriteten av studier sekvens endast en del av genen, eftersom den allmänt använda Illumina sekvenseringsplattform (högre genomströmning, lägre kostnad, minskad ansträngning jämfört med Sanger) producerar korta sekvenser ( ≤ 300 baser)., Olika subregioner av genen är därför riktade, allt från enstaka variabla regioner, såsom V4 eller V6, till tre variabla regioner, såsom V1–V3 eller V3–V5 (används i Human Microbiome projektet i samband med 454 sekvenseringsplattform9).
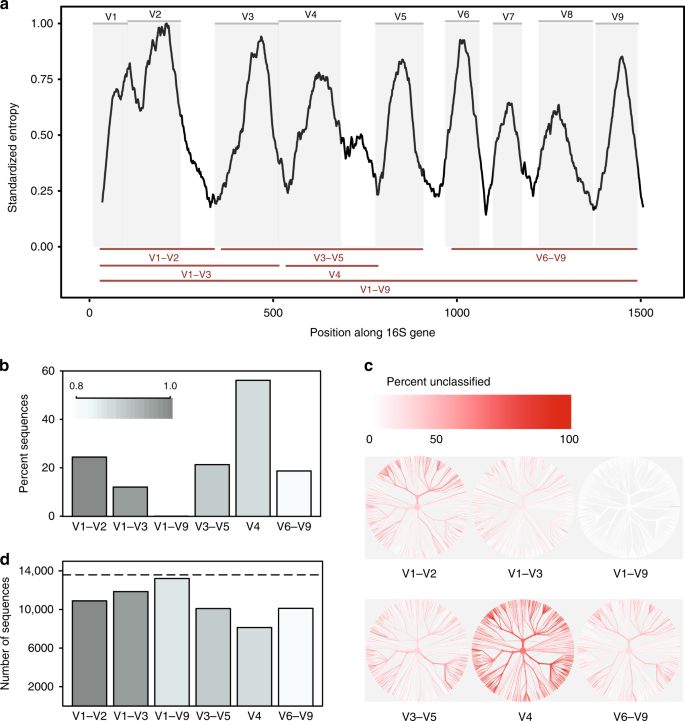
in-silico jämförelse av 16S rRNA variabla regioner. en Shannon entropi över 16S-genen baserad på anpassningen av en enda representativ sekvens för varje känd art som finns i Greengenes-databasen., Sekvenser jämfördes mot en enda referens genen för 16S Escherichia coli-K-12 MG1655 (NCBI Gen ID 947777). Grå paneler skildrar variabla områden som definieras av vanligen använda primerbindningsställen (Tilläggstabell 1). Variabla regioner som beaktas i denna studie visas som röda linjer (botten). b andel sekvenser för varje variabel region som inte kunde identifieras på artnivå när de klassificerade varje sekvens mot den referensdatabas från vilken den härleddes med en konfidenströskel på 80% (RDP-klassificerare)., C träd baserade på taxonomi av sekvenser som finns i in-silico databasen. Samma träd finns för varje variabel region. Färgen på varje gren återspeglar andelen sekvenser inom varje clade som inte kunde identifieras till artnivå. d antalet otu-enheter som skapas vid klustringssekvenser för varje variabel region vid 99% sekvens likhet. Streckad linje anger antalet unika sekvenser (>1% olika) i den ursprungliga databasen., Källdata tillhandahålls som en Källdatafil
Vi hävdar att inriktning av delregioner utgör en historisk kompromiss på grund av teknikbegränsningar10. Idag kan både PacBio och Oxford Nanopore-sekvenseringsplattformar rutinmässigt producera läser över 1500 bp och hög genomströmning sekvensering av hela 16S-genen blir allt vanligare., Vi föreslår därför att motiveringen till denna kompromiss måste ses över och vi utförde ett enkelt in-silico-experiment för att visa fördelen med fullängds 16S-sekvensering över inriktningen av delregioner.
vi hämtade en uppsättning icke-redundanta (dvs > 1% olika), fullängds 16S sekvenser från en offentlig databas (Greengenes)., Med fördel av det faktum att en betydande del av dessa sekvenser införlivade PCR primer-bindande platser, vi trimmade dem för att generera in-silico amplicons för olika delregioner, baserat på placeringen av PCR primers som vanligen används i mikrobiome studier (Fig. 1a och Tilläggstabeller 1-2)., Om vi antar att varje sekvens i vår nedladdade databas utgjorde en unik art använde vi sedan en gemensam klassificeringsmetod (ribosome Database Project (RDP) classifier11) för att beräkna frekvensen med vilken in-silico amplicons för varje delområde kunde ge korrekt, artnivå taxonomisk klassificering (med hjälp av den ursprungliga databasen som referens). I ett andra experiment, vi grupperade också våra In-silico amplicons att generera Otus vid olika, vanligen använda, sekvens likhet trösklar (97%, 98%, 99%).,
Vi fann att delregionerna skilde sig väsentligt i den utsträckning i vilken de med säkerhet kunde skilja mellan de 16-tals sekvenser i full längd som används för att representera arter (Fig. 1b). V4-regionen presterade värst, med 56% av in-silico amplicons som inte säkert matchar deras ursprungssekvens på denna taxonomiska nivå. När däremot en fullängdssekvens med alla variabla regioner användes, var det möjligt att klassificera nästan alla sekvenser som rätt Art (kompletterande Fig. 1a)., Genom att ändra databaser och klassificeringströsklar påverkades andelen in-silico-ampliconer som kunde matchas korrekt, men påverkade inte rådande trender (kompletterande Fig. 1a, B).
För det andra visade olika delregioner bias i den bakteriella taxa som de kunde identifiera (Fig. 1c). Till exempel, V1–V2 regionen utförs dåligt vid klassificering sekvenser som hör till fylum Proteobacteria, medan V3-V5 regionen utförs dåligt vid klassificering sekvenser som hör till fylum Aktinobacteria (kompletterande Fig. 2)., Liknande trender sågs på släkt nivå för taxa av potentiell medicinsk relevans. Även om hela V1–V9–regionen konsekvent gav de bästa resultaten, var V6-V9–regionen särskilt den bästa delregionen för att klassificera sekvenser som hör till genera Clostridium och stafylokocker, V3–V5-regionen gav bra resultat för Klebsiella, och V1-V3-regionen gav bra resultat för Escherichia/Shigella (kompletterande Fig. 2 och källdata).
slutligen påverkade valet av underregion dramatiskt antalet Otus som bildades vid klustring av in-silico amplicons för att skapa Otus., När gruppering på 99% sekvensidentitet, alla delregioner misslyckades med att återskapa antalet distinkta sekvenser som finns i den ursprungliga databasen; men v4-regionen återigen utfört värsta (Fig. 1d). Framför allt var det relativa antalet otu-enheter som producerades av varje delregion inte konsekvent vid olika identitetströsklar (97%, 98%, 99%, kompletterande Fig. 3), vilket indikerar att beteendet hos klusterningsalgoritmer kan vara svårt att förutsäga när mängden information som finns i en sekvenserad region är mycket varierande.,
Sammanfattningsvis utgör inriktning på delregioner en historisk kompromiss som var tillräcklig för identifiering av taxa på släkt nivå eller högre. Men vårt enkla in-silico-experiment visar att det inte är giltigt att anta att allt finare klustring av dessa underregioner kommer att resultera i den förbättrade taxonomiska upplösningen som är nödvändig för att återspegla arter. Även om vissa delregioner (t. ex. V1-V3) ger en rimlig approximation av 16S mångfald, fångar de flesta inte tillräcklig sekvensvariation för att diskriminera mellan närbesläktade taxa., Vi noterar också att diskriminerande polymorfism kan begränsas till specifika rörliga regioner.därför kommer vissa delregioner att vara bättre lämpade för att diskriminera nära besläktade medlemmar i vissa taxa.
16S genkopivarianter återspeglar stamnivåvariation
klustring av 16S-sekvenser i OTUs har historiskt tjänat två syften. För det första har det tagit bort mindre artefaktuella sekvensvarianter på grund av PCR-förstärkning och sekvenseringsfel vid kollapsande sekvenser i grupper. För det andra har det kollapsat legitima sekvensvarianter som finns mellan närbesläktade bakteriella taxa., Även om det senare kanske inte alltid är önskvärt, står det för att du inte kan skilja mellan bakteriell taxa vars 16S sekvenser varierar i en takt som är lägre än det fel som uppstått på en viss sekvenseringsplattform.
nyligen har framsteg inom CCS dramatiskt förbättrat felfrekvenserna för långlästa sekvenseringsplattformar. Samtidigt har beräkningsmetoder gjort det möjligt att skilja mellan legitim vs. artefaktisk sekvensvariation., Dessa tekniska och metodologiska framsteg innebär att forskare nu har potential att utföra hög genomströmning sekvensering som exakt kan upptäcka singelnukleotidvarianter över hela 16S-genen.
även om det är frestande att anta att singelnukleotidvarianter kan representera distinkta, närbesläktade taxa, varnar vi mot denna alltför förenklade Tolkning på grund av att många bakteriella genomer innehåller flera polymorfa kopior av 16S gene12,13,14., Vi utförde PacBio CCS-sekvensering av en 36 arter bakteriell mock gemenskapen (Kompletterande Tabell 3 och Kompletterande Fig. 4) för att visa (i) att 16S-sekvensen av många bakterier varierar mellan operoner inom samma genom och (ii) att hög genomströmning sekvensering är tillräckligt exakt för att lösa dessa intragenomiska skillnader.
Vi inriktade pacbio fullängds 16S sekvenser till en referensdatabas som innehåller en enda representant 16S sekvens för varje medlem i vår mock community och använde justeringsstatistiken för att utvärdera noggrannheten i denna sekvenseringsmetod., Att jämföra antalet passerkort som används för att generera en CCS med förekomsten av single-nucleotid substitutioner, Infogningar och deletioner indikerade att tio passerkort kan minimera dessa kombinerade fel till en minsta frekvens på< 1,0% (även om det var anmärkningsvärt att det minsta uppnåeliga felet varierade mellan sekvenseringskörningar; kompletterande Fig. 5). Men vi observerade en tillfällighet av radering fel med platsen homopolymer körs i våra referenssekvenser (kompletterande Fig., 6), som inte var nukleotidspecifik och förvärrades av längden på den sekvenserade homopolymeren (kompletterande Fig. 7). Vi validerade därefter deletioner inom Escherichia coli 16S-genen med Illumina whole genome shotgun (WGS) – sekvensering, vilket visade att endast en av de deletioner som förekommer i PacBio-sekvenser var äkta (kompletterande Fig. 8).,
övertygad om att CCS sekvensering kan producera 16S läser med en låg frekvens av substitutionsfel, vi nästa motiverade att en del av substitutionsfel inom noggrant inriktade läsningar bör återspegla variation hänförlig till 16S polymorfism inom en art ” genom12. Till exempel läser i linje med E. coli Stam K-12 substr. MG1655 visade en substitutionsprofil, som speglade exakt det som förutspåddes genom att anpassa alla sju av 16S-sekvenserna som är kända för att vara närvarande i detta genom15 (Fig. 2a, c)., Vi kunde vidare validera stökiometrin hos dessa nukleotidsubstitutioner genom att kvantifiera variation i jämförelsevis inriktade Illumina WGS läser (Fig. 2b) och visa att en liknande substitutionsprofil var reproducerbar över flera sekvenser (kompletterande Fig. 9)., Anpassningar till andra referenssekvenser i vårt mock-samhälle visade en liknande trend av rikliga substitutioner lokaliserade till specifika baspositioner längs 16S-genen, även om vi noterar att signal-brusförhållandet ökade signifikant när 16S-genen i fråga hade färre än 100-inriktade läsningar (kompletterande Fig. 10).

polymorfismer i E. coli 16S rRNA gensekvenser. a positionen och frekvensen av substitutioner som förekommer i E., coli-stam K-12 MG1655 V1–V9 amplikoner genereras från vår mock gemenskapen och analyserats på PacBio RS II-plattform. b substitutionernas position och frekvens i avläsningar genererade från genomisk sekvensering av den isolerade E. coli-stammen K-12 MG1655 på Illumina MiSeq-plattformen. Förstorade regioner visar respektive positioner i anpassningen av alla sju 16S-gener som finns i E. coli K-12 MG1655-referensgenomet. 16S-sekvensen från rrnd operon (**) används som referens för all SNP-fasning. C den förväntade nukleotid substitutionsprofilen för E., coli K-12 MG1655 baserat på anpassning av de sju 16S-gensekvenserna som finns i referensgenomet. d den förväntade substitutionsprofilen för E. coli O157 Sakai baserat på anpassning av de sju 16S-gensekvenserna som finns i referensgenomet. Grå paneler skildrar variabla områden som definieras av vanligen använda primerbindningsställen (Tilläggstabell 1). Streckade linjer indikerar den förväntade andelen nukleotidsubstitutioner, eftersom det finns sju 16S genkopior inom varje genom., Källdata tillhandahålls som en Källdatafil
observationen att långläst sekvensering kan identifiera 16S polymorfism inom samma Genom har viktiga konsekvenser. För det första visar det att det inte är giltigt att anta att hög genomströmning sekvens läser olika av en eller några nukleotider representerar en distinkt taxa6, 16. Inom ett enda Genom kan två eller flera 16S sekvenser vara identiska, medan andra kan vara unika., På motsvarande sätt kan vissa homologa 16S loci behålla identisk sekvens mellan två närbesläktade stammar, medan andra kan ha avvikit vid en eller några nukleotidpositioner. I detta sammanhang bör varje gemenskapsnivå eller taxonomisk tolkning av 16S-data helst ta hänsyn till det faktum att den relativa mängden 16S-sekvenser som härrör från mycket närbesläktade taxa kommer att återspegla en linjär kombination av i) den frekvens med vilken varje unik sekvens representeras över genom och ii) den relativa överflöd av genomen för varje taxon.,
För det andra, även om intragenomisk 16S-sekvensvariation komplicerar analys på gemenskapsnivå, har den också potential att öka kraften hos 16S-genen för att diskriminera mellan närbesläktade taxa, eftersom det möjliggör sekvensbaserad jämförelse för att sträcka sig över flera divergerande loci. Till exempel finns det tillräcklig nukleotidvariation för att skilja E. coli-Stam K-12 MG1655 från den enterohemorragiska stammen O157 Sakai (Fig. 2c, d)., Således hävdar vi att, när det är lämpligt redovisat, är flera polymorfa 16S-kopior inte ett besvär att förbises, utan de kommer att göra det möjligt för 16S-genen att användas i mikrobiomanalys på stamnivå. Vi noterar också att kraften i intragenomic 16S-sekvensvariation för att diskriminera närbesläktade taxa sannolikt kommer att minska när partiella 16S-sekvenser används. Till exempel, SNPs skilja E. coli stammar K-12 MG1655 (Fig. 2c) från O157 Sakai (Fig. 2d) finns i variabla regioner V1, V2, V6 och V9.,
16S polymorfism kan lösas in vivo
mikrobiom samhällen är ofta komplexa, befintliga i olika biokemiska miljöer (t.ex. avföring, saliv, slem, etc.) och innehåller många hundratals unika taxa vars relativa överflöd spänner över ett brett dynamiskt omfång. Denna komplexitet är inte väl representerad i antingen in-silico eller mock samhällsexperiment. Vi utförde därför ett ytterligare experiment för att visa att sekvensering av hela 16S-genen samtidigt som vi står för intragenomic 16S SNPs kan lösa nära besläktade bakteriella taxa in vivo.,
Vi utförde pacbio CCS-sekvensering av V1-V9-regionen för fyra mänskliga avföringsprover som samlats in från friska vuxna volontärer. Som jämförelse sekvenserade vi V1 – V3-regionen med Illumina MiSeq och för att ge ett riktmärke för artnivå taxonomisk kvantifiering utförde vi metagenomic WGS (mWGS) – sekvensering med Illumina NextSeq. För att utvärdera i vilken utsträckning var och en av dessa sekvenseringsmetoder kan lösa närbesläktade taxa, fokuserade vi på släktet Bacteroides., Förutom att vara riklig i människans tarm är detta släkt mycket varierat, innehållande flera arter som kan utöva både goda och dåliga effekter på människors hälsa17. Det har också tidigare använts som modell taxon för att demonstrera nyttan av 16S-genen för högupplösande taxonomisk analys18.
När vi beräknade Bacteroides överflöd på släktet nivå, V1–V9 sekvensering och V1–V3 sekvensering gav jämförbara resultat., Båda metoderna identifierade två individer med låga Bacteroides relativa överflöd (~10-25%) och två individer med höga Bacteroides relativa överflöd (~40-60%; Fig. 3a). Kvantifiering på artnivå via mwgs-sekvensering visade emellertid en mycket större mångfald, med en annan Bakterioidsart som dominerar i varje individs tarm (Fig. 3b och kompletterande uppgifter 1). Vid klustring av Otus vid 99% identitet kunde både V1-V9 och V1–V3-sekvensering återspegla denna artnivåvariation (Fig., 3b), med det anmärkningsvärda undantaget att V1–V3-sekvensering inte detekterade Bacteroides intestinalis, vilket var rikligt i ett av de fyra humana tarmmikrobiomeproverna. Baserat på dessa resultat drar vi slutsatsen att när de används tillsammans med en lämplig identitetströskel (t.ex. 99%) har otu-baserade metoder potential att lösa artnivådiversitet som observeras i den mänskliga tarmen. Vi noterar vidare att även om fullängds 16S-sekvensering kan vara optimal för artsnivåanalys, kan mycket informativa variabla regioner (t.ex. V1-V3) också vara tillräckliga för detta ändamål.,

detekterar Bakterioder i humana avföringsprover. A den relativa överflöd av släktet Bacteroides i fyra mänskliga avföringsprover kvantifieras med antingen V1-V9 amplicons (x-axel) eller V1–V3 amplicons (y-axel). b den relativa överflöd av Bakterieider arter i samma fyra prover. Art överflöd kvantifierades från mwgs sekvensering eller från V1-V3 / V1-V9 Otus genereras vid 99% identitet., Överflöd visas för de vanligaste arterna som kvantifieras av mWGS (för överflöd uppskattningar av alla Bakterieider arter som upptäcks av varje plattform, se kompletterande Tabell 5). C nukleotid substitutionsprofiler som genereras genom att anpassa alla V1–V9 ampliconsekvenser som tilldelats den enda otu som identifierats som Bacteroides vulgatus. Profiler visas för de två pallproverna med hög B. vulgatus relativ överflöd (IronHorse och Scott). d Nukleotid substitution profiler beräknas från referens arvsmassan hos två olika B. vulgatus stammar ATCC 848239 och mpk40., I både C och d identifierades nukleotidsubstitutioner i förhållande till en enda referens 16S-gen för B. vulgatus ATCC 8482 (NCBI-Gen ID 5304800). Grå paneler skildrar variabla områden som definieras av vanligen använda primerbindningsställen (Tilläggstabell 1). Streckade linjer indikerar den förväntade andelen nukleotidsubstitutioner, eftersom det finns sju 16S genkopior inom varje genom., Källdata tillhandahålls som en Källdatafil
med fördel av det faktum att Bacteroides vulgatus var närvarande vid hög relativ överflöd i två av våra mänskliga tarmmikrobiomeprover frågade vi nästa om intragenomisk variation mellan 16S-genkopior kunde detekteras in vivo. Vi inriktade varje Full längd sekvens klassificeras som tillhör vår B. vulgatus V1-V9 OTUs (Fig. 3b och kompletterande Data 1) till en enda representativ B. vulgatus 16S gensekvens. Vi jämförde sedan de resulterande nukleotid substitutionsprofilerna (Fig., 3C) med profiler som förutses från två referensgenom som finns i NCBI RefSeq database19 (Fig. 3d).
majoriteten av nukleotidvariationen i vår in vivo genererade B. vulgatus otu återspeglade sann variation hänförlig till intragenomiska polymorfism. Däremot verkade variationen sannolikt på grund av sekvenseringsfel låg och långt under den minimala ~ 14% – frekvensen som skulle förväntas om det fanns en enda B. vulgatus-stam i varje prov med sju 16S-genkopior i dess genom (Fig. 3C, streckade linjer).
även om vi inte visste det sanna antalet B., vulgatus-stammar som finns i varje In vivo-prov var det anmärkningsvärt att båda nukleotids substitutionsprofilerna hade närmare likhet med stam ATCC 8482 än mpk. Variation fanns också på specifika loci som potentiellt skulle kunna indikera meningsfulla skillnader mellan referensgenomen in vivo och ATCC 8482. Till exempel upptäcktes en enda polymorfism i V5-regionen ATCC 8482, som var närvarande i tre 16S-kopior (43%). I det första In-vivo-provet (Scott) var denna polymorfism närvarande i 84% av läsarna, medan den i den andra (IronHorse) var närvarande i 69% av läsarna., Dessa siffror motsvarar nära de siffror som förväntas om en polymorfism var närvarande sex och fem av sju 16S gener, respektive.
Sammanfattningsvis visar vi att fullängds 16S-sekvensering av den mänskliga tarmmikrobiomen kan exakt lösa singelnukleotidsubstitutioner som återspeglar intragenomisk variation mellan 16S-genkopior. Förekomsten av en sådan variation tyder på att 16S-sekvenser måste grupperas för att återspegla meningsfulla taxonomiska enheter., Med hjälp av Otus klustrade på 99% identitet, visar vi att full längd 16S har potential att ge arter och även stam-nivå taxonomisk upplösning. Analys av mikrobiella samhällen på dessa taxonomiska nivåer lovar att ge ett mycket annorlunda perspektiv till den som erbjuds av genus-nivå överflöd uppskattningar.
Intragenomiska 16S polymorfisms är mycket utbredd
Efter att ha visat att det är möjligt att lösa intragenomiska kopieringsvarianter in vivo, försökte vi nästa att fastställa i vilken utsträckning sådana kopieringsvarianter förekommer i taxa som vanligtvis finns inom den mänskliga tarmmikrobiomen., Vi försökte vidare fastställa om sådana profiler rutinmässigt kan användas för att skilja mellan stammar av samma art.
vi odlade 381 taxa från tarmmikrobiomen hos de friska individerna som avbildas i Fig. 3, liksom från andra personer som deltar i samma ursprungliga studie20 (kompletterande uppgifter 2). Vi utförde därefter fullängds 16S gensekvensering på isolat och inriktade sekvenserade läsningar för att identifiera nukleotidsubstitutioner som är karakteristiska för intragenomiska 16S genkopivarianter.,
taxonomisk klassificering av isolat identifierade 58 putativa arter (kompletterande Data 2), medan klustring av en enda representativ sekvens för varje isolat vid 99% likhet resulterade i 61 Otus (med mellan 1 och 73 isolat tilldelade varje otu). Totalt hade 349 av 381 sekvenserade isolat (54 av 61 Otus) en eller flera SNP, vilket indikerar närvaron av 16S-genpolymorfismer och 205 unika SNP-profiler identifierades vid redovisning av potentiellt sekvenseringsfel (Fig. 4a och kompletterande uppgifter 2).

Intragenomiska 16S genpolymorfism i human tarmmikrobiome isolat. en plats av SNPs närvarande i 16S gener av individuellt odlade bakteriella isolat. SNP platser identifierades genom fasning Full längd 16S gensekvenser som genereras för varje enskilt isolat. X-axeln betecknar position längs 16S-genen. Y-axeln betecknar enskilda isolat grupperade baserat på deras härledda fylogeni. Mörkblå region indikerar placeringen av en polymorfism., För tydlighetens skull visas högst fem isolat som tillhör samma art. För mer information om nukleotid substitutionsprofiler för alla sekvenserade isolat, se kompletterande Data 2. B-D exempel på nukleotid substitutionsprofiler som visar skillnader på stamnivå mellan isolat som identifierats som tillhör tre bakteriearter: B Shigella flexneri; C Bifidobacterium longum; d Collinsella aerofaciens. För varje art visas två isolat nukleotid substitutionsprofiler, men ytterligare exempel finns i kompletterande Data 2., Isolat identifierades tillhöra samma art om deras representativa sekvenser tilldelades samma OTU vid klustring vid 99% sekvensidentitet. Taxonomisk identifiering utfördes med BLAST för att anpassa representativa sekvenser till NCBI 16S BLAST Databas (se metoder). Grå paneler skildrar variabla områden som definieras av vanligen använda primerbindningsställen (Tilläggstabell 1). Streckade linjer indikerar den förväntade andelen nukleotidsubstitutioner, med tanke på antalet 16S-genkopior som förutses för varje genom., Källdata tillhandahålls som en Källdatafil
genom att jämföra SNP-profiler för isolat som tilldelats samma OTU avslöjade ofta skillnader i frekvensen av SNPs som tydde på skillnader i intragenomiska 16S-genkopior mellan närbesläktade taxa. Exempel på olika substitutionsprofiler visas för tre taxa (Fig. 4b-d), som är suggestiva av stamnivåvariation jämförbar med den som vi i princip visade för E. coli (Fig. 2b).,
Sammanfattningsvis visar vi att många av de odlingsbara medlemmarna i den mänskliga tarmmikrobiomen ofta har 16S-genpolymorfismer, som, när de är korrekt redovisade, har potential att lösa stammar av samma art.